Malattia di Hirschsprung
Astrid Leitner ha studiato medicina veterinaria a Vienna. Dopo dieci anni di pratica veterinaria e la nascita della figlia, è passata, più per caso, al giornalismo medico. Divenne subito chiaro che il suo interesse per gli argomenti medici e il suo amore per la scrittura erano la combinazione perfetta per lei. Astrid Leitner vive con figlia, cane e gatto a Vienna e nell'Alta Austria.
Maggiori informazioni sugli esperti di Tutti i contenuti di sono controllati da giornalisti medici.La malattia di Hirschsprung è una malattia congenita in cui le cellule nervose mancano nell'ultima sezione del colon. Le persone affette spesso soffrono di stitichezza permanente fino all'ostruzione intestinale dalla nascita. Nella maggior parte dei casi, la chirurgia è essenziale. Leggi tutto quello che c'è da sapere sulla prognosi, i sintomi e il trattamento del "megacolon congenito" qui!
Codici ICD per questa malattia: i codici ICD sono codici riconosciuti a livello internazionale per le diagnosi mediche. Si trovano, ad esempio, nelle lettere dei medici o nei certificati di inabilità al lavoro. Q43

Breve panoramica
- Che cos'è la malattia di Hirschsprung?: Malformazione congenita della sezione più bassa dell'intestino crasso
- Prognosi: con un trattamento tempestivo e controlli regolari, la prognosi è buona.
- Sintomi: nausea, vomito, gonfiore, ritardata o assente emissione delle prime feci nel neonato (“Kindspech”), costipazione, ostruzione intestinale, dolore addominale
- Cause: mancanza di cellule nervose nell'ultima sezione del colon
- Fattori di rischio: trisomia 21 (o altri rari difetti cromosomici), predisposizione genetica
- Diagnostica: sintomi tipici, esame obiettivo, raggi X, ultrasuoni, misurazione della pressione nel retto, biopsia, esame genetico
- Trattamento: nella maggior parte dei casi, è necessario un intervento chirurgico.
- Prevenzione: la malattia di Hirschsprung è una malattia congenita, la prevenzione non è possibile.
Cos'è la malattia di Hirschsprung?
La malattia di Hirschsprung (MH), nota anche come megacolon congenito, è una malformazione congenita che colpisce l'ultima sezione dell'intestino crasso e lo sfintere interno all'ano. In questi punti mancano le cellule nervose che normalmente controllano i muscoli intestinali. I muscoli intestinali assicurano che il contenuto intestinale venga trasportato ulteriormente verso il retto. Per fare ciò, i muscoli si contraggono in modo ondulatorio e quindi spingono ulteriormente il contenuto intestinale. I medici chiamano questo processo peristalsi.
Nei pazienti con MH, la peristalsi è disturbata a causa della mancanza di cellule nervose. Il punto in cui mancano i nervi è permanentemente ristretto (stenosi funzionale). A seconda di quanto è grande la sezione dell'intestino interessata, il contenuto dell'intestino non viene trasportato ulteriormente o solo in quantità molto piccole.
Le feci si accumulano sempre più davanti al punto ristretto e dilatano la sezione di intestino davanti ad esso. Questa grande espansione dell'intestino crasso è anche conosciuta come megacolon. Trattandosi di una malattia congenita, i medici parlano anche di "megacolon congenito" o "megacolon congenitum". Il nome della malattia di Hirschsprung risale al pediatra Harald Hirschsprung, che per primo descrisse la malattia nel 1888.
Se l'intestino non si svuota completamente, si accumulano sempre più feci. Nella sezione allungata dell'intestino piena di feci, può svilupparsi un'infiammazione (enterocolite), nel peggiore dei casi un'ostruzione intestinale. Se non trattata, ciò alla fine porta ad avvelenamento del sangue (sepsi) o a una crescita eccessiva batterica pericolosa per la vita dell'intestino crasso (megacolon tossico).
La durata della sezione dell'intestino interessata nella malattia di Hirschsprung varia da paziente a paziente. È tipico, tuttavia, che la mancanza di cellule nervose inizi sempre nell'ano e si estenda verso l'alto a diversi gradi nell'intestino. Poiché anche l'ano è interessato, il muscolo dello sfintere interno non ha la capacità di rilassarsi, il che rende ancora più difficile la defecazione. Il pezzo di intestino non funzionante viene anche chiamato segmento privo di cellule nervose (agangliari), motivo per cui i medici parlano anche di aganglionosi.
frequenza
La malattia di Hirschsprung è una delle malattie rare. Ogni anno ne nasce circa un bambino su 5.000. I ragazzi hanno circa quattro volte più probabilità di essere colpiti rispetto alle ragazze.
Testo di avvio (teaser): La malattia di Hirschsprung è una malattia congenita in cui le cellule nervose mancano nell'ultima sezione del colon. Le persone affette spesso soffrono di stitichezza permanente fino all'ostruzione intestinale dalla nascita. Nella maggior parte dei casi, la chirurgia è essenziale. Leggi tutto quello che c'è da sapere sulla prognosi, i sintomi e il trattamento del "megacolon congenito" qui!
Qual è l'aspettativa di vita dei pazienti con MH?
Se la malattia di Hirschsprung viene trattata in tempo utile, la prognosi è buona e l'aspettativa di vita non è limitata. Tuttavia, la malattia richiede cure di follow-up a lungo termine e controlli regolari per tutta la vita, anche dopo un'operazione riuscita. Nella maggior parte dei casi, la chirurgia può ripristinare completamente la funzione intestinale.
Sintomi
I sintomi che si verificano dipendono dalle dimensioni della sezione dell'intestino interessata e dal numero di cellule nervose mancanti. Nella maggior parte dei casi, i sintomi tipici compaiono nei primi giorni di vita.
I sintomi tipici nei bambini sono:
Il meconio ("Kindspech") è il primo sgabello in un neonato. Non è un classico movimento intestinale, ma un accumulo di cellule della pelle e delle mucose, succhi digestivi e liquido amniotico ingerito. Il meconio è tipicamente nero-verdastro, inodore e viene escreto entro i primi due giorni di vita.
Se il meconio non scompare dopo il secondo giorno, potrebbe trattarsi della malattia di Hirschsprung. Nei neonati, anche un addome notevolmente spesso e disteso può indicare MH.
I sintomi tipici nei bambini sono:
Se è interessata solo una breve sezione dell'intestino, i genitori potrebbero non notare immediatamente la malformazione. I movimenti intestinali modificati sono spesso evidenti solo dopo il periodo dell'allattamento al seno. Il motivo: finché i bambini sono allattati al seno, le loro feci sono relativamente sottili e quindi più facili da trasportare. A volte non si nota una costrizione nell'intestino. I problemi sorgono solo quando si cambia il cibo. I bambini affetti possono sviluppare i seguenti sintomi e condizioni:
- Feci dure o pastose, pastose, occasionalmente anche liquide
- Costipazione costante, movimenti intestinali solo ogni due o tre giorni
- Evacuazione delle feci possibile solo con ausili (clistere, inserimento delle dita o termometro)
- Le feci hanno un cattivo odore
- mal di stomaco
- Vomito
- Rifiuto di mangiare
- Cattive condizioni generali
Cause e fattori di rischio
La causa della malattia di Hirschsprung è la mancanza di cellule nervose nell'intestino crasso. Il contenuto intestinale viene a malapena o non più trasportato nelle aree colpite e si accumula. A seconda del numero di cellule nervose mancanti, vengono colpite sezioni dell'intestino crasso di diversa lunghezza.
cause
La ragione della mancanza di cellule nervose è uno sviluppo embrionale disturbato tra la quarta e la dodicesima settimana di gravidanza. Come ciò avvenga non è del tutto chiaro. I medici presumono che diversi fattori svolgano un ruolo:
- Cause genetiche: la malattia di Hirschsprung è una malattia che si verifica nelle famiglie. Il rischio di avere un figlio con MH dopo aver avuto un altro figlio con la stessa malattia è del 4%. Fondamentalmente, più lungo è il pezzo di intestino privo di cellule nervose, maggiore è il rischio di trasmettere la malformazione alla sua prole.
- Ridotto flusso sanguigno all'embrione
- Infezioni virali nell'utero
- Disturbo della maturazione
Fattori di rischio
La maggior parte delle volte, la malattia di Hirschsprung si presenta come una malattia indipendente. I bambini nati con la trisomia 21 ("sindrome di Down"), tuttavia, hanno un aumentato rischio di malattia di Hirschsprung: circa un terzo dei pazienti con MH è associato alla trisomia 21 o ad altre malattie genetiche come la sindrome di Undine, la sindrome di Shah Waardenburg o la sindrome di Mowat Wilson .
diagnosi
Nella maggior parte dei casi, i primi sintomi compaiono durante l'infanzia. Il pediatra è il primo punto di contatto se si sospetta MH. L'anamnesi ei sintomi tipici danno le prime indicazioni della malattia di Hirschsprung. Il pediatra conferma la diagnosi con ulteriori esami:
Esame fisico: il medico prima sente lo stomaco e presta attenzione ai cambiamenti tipici come stomaco gonfio o feci dure palpabili. Effettua quindi un esame "digito-rettale": esamina con il dito l'ano e il retto. Un retto stretto che non contiene feci può essere un segno della malattia di Hirschsprung. Anche i rumori intestinali chiaramente udibili indicano la malattia intestinale.
Esame ecografico: un esame dell'addome mediante ultrasuoni consente al medico di rilevare l'allargamento o un forte riempimento dell'intestino con le feci. Può anche vedere se l'intestino si sta muovendo (peristalsi intestinale).
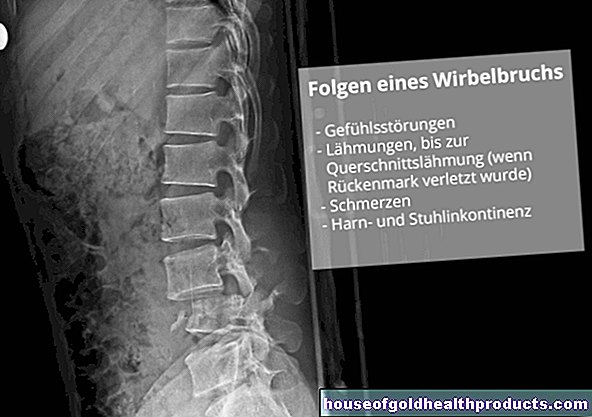
Esame a raggi X dell'intestino crasso (raggi X del colon): per vedere se l'intestino è ristretto in una certa area, il medico inietta un mezzo di contrasto nell'intestino attraverso un tubo sottile. Se nella radiografia si riscontrano costrizioni a forma di imbuto, questa è un'indicazione della malattia di Hirschsprung.
Rimozione del tessuto dalla parete intestinale (biopsia): dalla sesta settimana di vita è possibile una colonscopia con rimozione simultanea del tessuto (biopsia). Per fare ciò, il medico preleva un campione di tessuto dalla parete intestinale e utilizza un microscopio e test speciali per esaminare se sono presenti cellule nervose. La diagnosi viene fatta quando non ci sono (o sono troppo poche) cellule nervose nella parete intestinale.
Misurazione della pressione rettale (manometria rettale): durante questa visita il medico controlla se c'è peristalsi e se i due sfinteri anali funzionano. Nel caso della MH, la parete intestinale si contrae come uno spasmo, i muscoli dello sfintere sono strettamente chiusi. Importante: la manometria rettale ha un significato limitato per neonati e bambini, poiché questo metodo di esame richiede la collaborazione del paziente.
Diagnostica genetica molecolare: circa un terzo dei pazienti con MH è affetto contemporaneamente da trisomia 21 (o altri rari difetti cromosomici). Questi possono essere determinati utilizzando metodi genetici molecolari (esami del sangue), ma non i cambiamenti genetici che scatenano l'MH. Ad oggi, questi non sono stati ancora completamente esplorati.
trattamento
Nella maggior parte dei casi di malattia di Hirschsprung, è necessario un intervento chirurgico. Quando la procedura viene eseguita dipende principalmente dalle condizioni generali del paziente.
Intervento chirurgico immediato se le condizioni generali sono buone
I pazienti con MH vengono solitamente operati presto in modo che non ci siano complicazioni come un'ostruzione intestinale. Durante l'operazione, il chirurgo rimuove la sezione interessata dell'intestino e quindi anche la costrizione, in modo che siano nuovamente possibili i normali movimenti intestinali. Il chirurgo esegue la procedura attraverso l'ano o attraverso la cavità addominale.
Misure ponte fino all'intervento se le condizioni generali sono scadenti
Se le condizioni generali sono scarse (neonato debole) o se si sono già verificate complicazioni come avvelenamento del sangue (sepsi), potrebbe essere necessario attendere un po' prima dell'operazione. In questi casi, il medico rimuove regolarmente le feci fino all'operazione sciacquando l'intestino (clistere) o allargando la sezione intestinale con strumenti speciali.
A volte è necessario creare temporaneamente un ano artificiale (stoma) prima dell'operazione vera e propria.
Dopo l'operazione
Occorrono alcune settimane prima che la ferita chirurgica nell'intestino guarisca e che l'intestino crasso torni a funzionare normalmente. Durante questo periodo, i genitori dei bambini affetti devono osservare i seguenti punti:
- Frequenti movimenti intestinali: molti bambini hanno movimenti intestinali molto frequenti (da cinque a 30 volte) e sottili dopo l'operazione. La pelle può reagire in modo molto sensibile a questo. Affinché il fondoschiena non si irriti, si consiglia di prendersi cura della pelle particolarmente (soprattutto nella zona del pannolino).
- Farmaci: nelle prime settimane dopo l'operazione, ai bambini vengono somministrati farmaci che influiscono sulla consistenza delle feci. È importante che i bambini affetti prendano regolarmente il farmaco.
- Prevenzione delle cicatrici: dopo l'operazione, è necessario espandere il sito chirurgico nell'intestino in modo che il sito non si cicatrizzi e quindi si restringa di nuovo. Questo viene fatto con spilli speciali che allungano il tessuto.
- Se è stato creato un ano artificiale, verrà rimosso di nuovo dopo tre o quattro settimane.
Prevenire
Poiché la malattia di Hirschsprung è una malattia congenita, non esistono misure per prevenire la malattia.
Tags.: Malattie menopausa anatomia