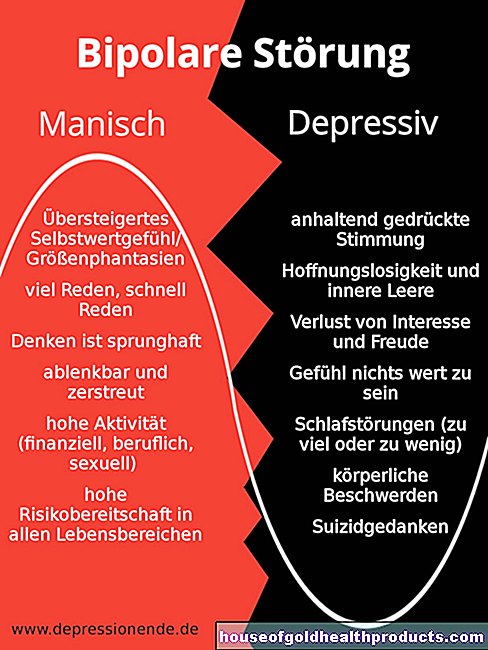
Questioni regolamentari
Martina Feichter ha studiato biologia con una materia elettiva farmacia a Innsbruck e si è anche immersa nel mondo delle piante medicinali. Da lì non era lontano da altri argomenti medici che la affascinano ancora oggi. Si è formata come giornalista presso l'Axel Springer Academy di Amburgo e lavora per dal 2007 - prima come redattrice e dal 2012 come scrittrice freelance.
Maggiori informazioni sugli esperti di Tutti i contenuti di sono controllati da giornalisti medici.
La ricerca di nuovi principi attivi contro determinate malattie o disturbi è tediosa e non sempre finisce con successo. Su 5.000 a 10.000 aspiranti che vengono testati nei laboratori di ricerca delle aziende farmaceutiche, in media solo uno finisce come farmaco finito in farmacia. E c'è una media di 13,5 anni nel mezzo.

Cerca il "bersaglio"
Ancor prima che vengano effettuati test con nuove sostanze, i ricercatori pensano alle proprietà della sostanza in questione e a quale reazione dovrebbe innescare nell'organismo. Questo può essere, ad esempio, l'abbassamento della pressione sanguigna, il blocco di una determinata sostanza messaggera o il rilascio di un ormone.
A tal fine, i ricercatori stanno cercando un "bersaglio" adatto, cioè un punto di attacco nel processo della malattia, in cui un ingrediente attivo può attaccarsi e quindi influenzare positivamente il processo della malattia. Nella maggior parte dei casi il bersaglio è un enzima o un recettore (punto di attracco sulle cellule per ormoni o altre sostanze messaggere). A volte al paziente manca anche una certa sostanza. Quindi diventa subito chiaro che il farmaco che stai cercando dovrebbe compensare questa carenza. Un esempio ben noto è l'insulina nelle persone con diabete (diabete mellito).
Cerca il principio attivo
Non appena viene determinato un obiettivo, gli scienziati cercano un ingrediente attivo che possa agire sul punto di attacco prescelto (screening). Questo di solito significa: prova, prova, prova. Ogni giorno vengono esaminate fino a 300.000 sostanze diverse per verificarne l'idoneità (screening ad alto rendimento = HTS). Di questi, circa ogni 200-1000 sostanza mostra effettivamente un effetto sul target selezionato, anche se a volte solo molto piccolo. Tale colpo è indicato come un "colpo".
Le sostanze di prova sono per lo più prodotte chimicamente, ovvero sinteticamente. Da qualche tempo stanno acquistando importanza anche le sostanze geneticamente modificate. Sono ottenuti con l'aiuto di cellule geneticamente modificate (come alcuni batteri) e costituiscono la base dei biofarmaci (farmaci biologici).
ottimizzazione
Nella maggior parte dei casi, gli "hit" trovati devono ancora essere ottimizzati. A volte, ad esempio, l'efficacia di una sostanza può essere aumentata se la sua struttura viene leggermente modificata. In questi esperimenti, gli scienziati lavorano spesso con simulazioni al computer, con l'aiuto delle quali è possibile stimare in anticipo l'effetto di un cambiamento chimico sulla sostanza. Se la prognosi è buona, la sostanza viene adattata nella vita reale, cioè in laboratorio. Il loro effetto sul bersaglio viene quindi esaminato di nuovo.
In questo modo, i ricercatori migliorano gradualmente un nuovo principio attivo, che di solito richiede diversi anni.Nel migliore dei casi, alla fine raggiungeranno il punto in cui la sostanza è pronta per il passaggio successivo: viene depositata una domanda di brevetto e quindi sottoposta a studi preclinici come un cosiddetto principio attivo candidato.
Studi preclinici
Nella fase di sviluppo preclinico (preclinico), il farmaco candidato viene testato in provette (ad es. su colture cellulari) e su animali. Da un lato, ciò comporta questioni farmacologiche, ad esempio cosa accade alla sostanza nelle cellule o in un intero organismo:
- Come viene ricevuto?
- Come si distribuisce nel corpo?
- Quali reazioni scatena?
- Verrà modificato o smantellato?
- Verrà eliminato?
D'altra parte, gli scienziati stanno studiando esattamente quale effetto ha la sostanza sul bersaglio, quanto dura e quale dose è necessaria per essa.
Soprattutto, tuttavia, gli studi preclinici servono a rispondere a domande sulla tossicità (tossicità) del candidato farmaco. La sostanza è velenosa? Può causare il cancro? È in grado di cambiare i geni? Può danneggiare un embrione o un feto?
Molti candidati ai farmaci non superano i test di tossicità. Solo quelle sostanze che superano tutti i test di sicurezza possono entrare nella successiva fase di sviluppo con studi sull'uomo (studi clinici).
Quando possibile, i test preclinici vengono eseguiti in provetta, ad esempio su colture cellulari, frammenti cellulari o organi umani isolati. Tuttavia, alcune domande possono essere risolte solo nei test su un organismo vivente e per questo sono necessari esperimenti sugli animali.
Studi clinici
Il candidato farmaco viene testato per la prima volta sull'uomo negli studi clinici. Viene fatta una distinzione tra tre fasi di studio che si basano l'una sull'altra:
- Fase I: il candidato al farmaco viene testato su alcuni volontari sani (soggetti del test).
- Fase II: questa è seguita da test su alcune persone malate (ad esempio su pazienti con pressione alta se il candidato al farmaco deve diventare un nuovo agente antipertensivo).
- Fase III: ora il test viene effettuato su un gran numero di malati.
Ogni fase dello studio deve essere preventivamente approvata dagli organismi responsabili: da un lato, ciò include l'autorità nazionale responsabile - a seconda del candidato al farmaco, l'Istituto federale per i farmaci e i dispositivi medici (BfArM) o il Paul Ehrlich Institute (PEI ). D'altra parte, ogni studio clinico necessita del permesso di un comitato etico (composto da medici, avvocati, teologi e laici). Questa procedura ha lo scopo di proteggere i partecipanti allo studio nel miglior modo possibile.
Il produttore farmaceutico che ha sviluppato il farmaco candidato può condurre personalmente gli studi clinici. Oppure assume un "Clinical Research Organization" (CRO) per farlo. Questa è una società specializzata nella conduzione di studi clinici.
Studi di fase I
Di solito da 60 a 80 adulti sani che si sono offerti volontari per questo atto come persone di prova nella fase I. Dopo una spiegazione esauriente e il consenso dei partecipanti allo studio, inizialmente viene data loro solo una piccola quantità di principio attivo.
In un massimo di 30 test consecutivi, gli scienziati verificano se i risultati dei test in provetta e sugli animali possono essere trasferiti anche all'uomo, ovvero se il principio attivo viene assorbito, distribuito, convertito ed escreto di nuovo come farebbe nella fase preclinica Prove determinate. Inoltre, viene studiato il modo in cui le persone del test tollerano il candidato al farmaco.
Compressa, siringa o unguento?
Dopo aver completato con successo la fase I, entra in gioco la cosiddetta galenica: gli scienziati stanno ora lavorando al "packaging" ottimale per il principio attivo: dovrebbe essere somministrato in vena come compressa, capsula, supposta, siringa o infusione?
La risposta a questa domanda è molto importante: la forma di dosaggio ha una grande influenza sull'affidabilità, la rapidità e la durata del principio attivo in grado di svolgere il suo compito nel corpo. Colpisce anche il tipo e la gravità dei possibili effetti collaterali. Alcuni principi attivi sono molto meglio tollerati come iniezione rispetto a quando entrano nel corpo sotto forma di compresse attraverso il tratto gastrointestinale.
Inoltre, gli esperti galenici verificano se e quali sostanze ausiliarie devono essere aggiunte al nuovo preparato. Ad esempio, qualcosa che migliora il gusto del farmaco o funge da vettore o conservante.
Puoi leggere di più sulla ricerca del "packaging" giusto per un nuovo ingrediente attivo e per i materiali ausiliari adeguati nell'articolo Galenics - Fabbricazione di prodotti farmaceutici.
Studi di Fase II e Fase III
Dopo i soggetti sani in fase I, è il turno dei malati della fase II di testare il candidato farmaco:
- Fase II: qui il nuovo candidato farmaco viene testato per lo più da 100 a 500 pazienti. L'attenzione è rivolta all'efficacia, al dosaggio ottimale e alla tollerabilità del preparato.
- Fase III: qui vengono eseguiti gli stessi controlli della Fase II, solo su un numero significativamente maggiore di pazienti (diverse migliaia). Inoltre, viene prestata attenzione alle possibili interazioni con altri farmaci.
In entrambe le fasi, vengono confrontati diversi trattamenti tra loro: solo alcuni dei pazienti ricevono la nuova preparazione, il resto riceve un farmaco standard abituale o familiare o un placebo - una preparazione che assomiglia esattamente a quella nuova ma non contiene alcun principio attivo (farmaco fittizio) . Di norma, né il paziente né il medico curante sanno chi riceve cosa. Tali "studi in doppio cieco" hanno lo scopo di evitare che speranze, paure o atteggiamenti scettici di medici e pazienti influenzino l'esito del trattamento.
Concessione dell'approvazione
Anche se un nuovo farmaco ha superato tutti gli studi e i test richiesti, non può essere semplicemente venduto. Per fare ciò, l'azienda farmaceutica deve prima richiedere l'approvazione del farmaco all'autorità competente (vedi sotto: Opzioni di approvazione). Questo controlla attentamente tutti i risultati dello studio e, nel migliore dei casi, concede al produttore il permesso di immettere il nuovo farmaco sul mercato.
Fase IV
Anche dopo che un farmaco è stato approvato, le autorità e l'azienda farmaceutica tengono d'occhio il nuovo preparato, ad esempio per quanto riguarda gli effetti collaterali rari. Questi sono effetti indesiderati che si verificano in meno di 1 paziente su 10.000 trattati e sono quindi difficilmente rilevabili nelle precedenti fasi dello studio (con gruppi di pazienti più piccoli). I medici sono tenuti a segnalare eventuali effetti collaterali imprevisti di un farmaco.
Se necessario, l'autorità di omologazione chiederà quindi al produttore di indicare questi effetti collaterali appena scoperti nel foglietto illustrativo. Tuttavia, può anche imporre restrizioni sull'uso: se, ad esempio, sono stati scoperti effetti collaterali rari ma gravi nell'area renale, le autorità possono ordinare che il farmaco non possa più essere utilizzato nelle persone con malattie renali esistenti.
In casi estremi, le autorità possono revocare del tutto l'approvazione di un farmaco se, nel tempo, sono emersi rischi inaccettabili dal suo uso. A volte il produttore ritira volontariamente un tale prodotto dal mercato.
I medici usano anche i registri per registrare come sta andando il nuovo farmaco nella vita di tutti i giorni per i loro pazienti. Il produttore utilizza i risultati di tali studi osservazionali, ad esempio, per migliorare il dosaggio o la forma di dosaggio del preparato.
A volte la pratica quotidiana mostra anche che il principio attivo aiuta contro altre malattie. Il produttore di solito continua la ricerca in questa direzione, con nuovi studi di fase II e III. In caso di successo, può anche richiedere l'approvazione per questa nuova indicazione.
Opzioni di approvazione
In linea di principio, un'azienda farmaceutica può richiedere l'approvazione di un nuovo farmaco per l'intera UE o solo per un singolo Stato membro:
Processo di approvazione centralizzato
L'approvazione del farmaco viene richiesta qui direttamente dall'Agenzia europea per i medicinali (EMA). Al test successivo partecipano anche le autorità di omologazione degli Stati membri dell'UE. Se la domanda viene approvata, il preparato può essere venduto ovunque nell'UE. Questo processo di approvazione richiede in media un anno e mezzo ed è obbligatorio per alcuni farmaci (ad es. per preparati fabbricati biotecnologicamente e per farmaci antitumorali con nuovi principi attivi).
Processo di approvazione nazionale
La domanda di approvazione è presentata alle autorità nazionali e quindi solo nel paese interessato. In Germania, ne sono responsabili l'Istituto federale per i farmaci e i dispositivi medici (BfArM) e il Paul Ehrlich Institute (PEI). Il BfArM si occupa della maggior parte dei prodotti farmaceutici umani, il PEI si occupa di sieri, vaccini, allergeni di prova, sieri di prova e antigeni di prova, sangue ed emoderivati, tessuti e farmaci per la terapia genica e cellulare.
Approvazione di farmaci in diversi paesi dell'UE
Inoltre, ci sono altre due opzioni se un'azienda farmaceutica vuole ottenere l'approvazione in diversi paesi dell'UE:
- Procedura decentralizzata: in una "procedura decentralizzata" (DCP), un'azienda farmaceutica può richiedere l'approvazione nazionale per un nuovo farmaco in più paesi dello Spazio economico europeo contemporaneamente.
- Procedura di mutuo riconoscimento: se un farmaco ha già un'approvazione nazionale in un paese dello Spazio economico europeo, questa può essere riconosciuta da altri Stati membri nell'ambito della "Procedura di mutuo riconoscimento" (MRP).
La domanda di approvazione per un nuovo farmaco è molto costosa per le aziende farmaceutiche. Ad esempio, l'elaborazione di una domanda di approvazione per un principio attivo completamente nuovo presso l'EMA costa circa 260.000 euro nel caso più semplice.
Approvazione standard
Alcuni farmaci vengono messi in vendita tramite un'approvazione standard: non si tratta di preparati di nuova concezione, ma di quelli la cui produzione si basa su determinate monografie stabilite dal legislatore. Inoltre, questi medicinali non devono rappresentare alcun pericolo per l'uomo o gli animali. In una monografia (ad es. per supposte di paracetamolo 250 mg), tra l'altro, la composizione e il dosaggio del preparato in questione sono definiti con precisione, così come l'area di applicazione.
Se tutti questi requisiti sono soddisfatti, il produttore non deve richiedere la propria approvazione individuale del farmaco. Ciò gli consente di immettere sul mercato farmaci a un prezzo molto conveniente. Esistono approvazioni standard per compresse di carbone (250 mg), colliri e soluzioni di atropina in varie concentrazioni, nonché supposte di paracetamolo e compresse di acido acetilsalicilico in vari dosaggi.
I farmacisti, ad esempio, possono anche preparare una soluzione salina secondo le istruzioni della farmacopea interessata e poi venderla. Tuttavia, è necessario indicare l'uso di tale approvazione standard all'autorità di approvazione e all'autorità statale responsabile.
Altri modi per ottenere l'approvazione dei farmaci
Nell'UE, oltre alla procedura di approvazione convenzionale, esistono anche opzioni per rendere disponibile un nuovo farmaco prima del solito. Queste non sono solo approvazioni rapide. Piuttosto, si stanno facendo tentativi in vari modi per garantire che le persone colpite possano beneficiare dei principi attivi anche senza l'approvazione dei farmaci tradizionali. Gli esperti parlano dei cosiddetti percorsi adattivi:
Programmi di uso compassionevole
Qui, pazienti molto specifici ricevono farmaci che sono ancora in fase di sperimentazione clinica. Il prerequisito è che non ci siano più altre opzioni di trattamento e il paziente non può partecipare a uno studio corrispondente su questo farmaco. Tali esenzioni devono essere richieste separatamente per ogni singolo paziente.
Approvazione condizionata per i medicinali
Si tratta, per così dire, di una rapida approvazione. I severi test di efficacia e sicurezza non devono essere presenti nella misura in cui è altrimenti normale. Tuttavia, si applicano alcune condizioni:
- L'approvazione condizionata del farmaco è limitata nel tempo.
- Il produttore deve fornire i documenti mancanti necessari per la regolare approvazione del farmaco
L'approvazione condizionata viene utilizzata, ad esempio, nelle pandemie per fornire rapidamente un farmaco adatto contro la malattia infettiva.
Approvazione in circostanze eccezionali
Questo percorso speciale è disponibile, ad esempio, per le malattie rare. Poiché ci sono pochissimi malati, non è possibile per l'azienda farmaceutica presentare la quantità di dati altrimenti necessaria per l'esame. Con questa approvazione del farmaco, tuttavia, il produttore di solito deve controllare annualmente se ci sono nuovi dati e scoperte.
Approvazione accelerata dei farmaci (valutazione accelerata)
I documenti di approvazione vengono controllati e valutati più rapidamente dal comitato EMA responsabile, invece dei soliti 210 in 150 giorni. Questa strada è possibile se esiste un promettente ingrediente attivo contro una malattia che fino ad ora non è stata curata adeguatamente.
Medicinali prioritari (PRIME)
In questi casi in cui un'esigenza non è ancora soddisfatta, l'EMA e il produttore del farmaco possono collaborare molto presto, anche durante i primi test. In questo modo, gli esperti possono valutare l'efficacia e la sicurezza in una fase iniziale e avviare ulteriori procedure più rapidamente se il farmaco si dimostra promettente.
Revisione continua (revisione continua)
Nel caso di farmaci e vaccini urgenti, l'EMA può - come già notato - approvare "condizionatamente" i principi attivi o collaborare con i produttori in una fase iniziale prima dell'approvazione finale. In casi importanti, il cosiddetto processo di revisione continua inizia prima di queste approvazioni. Gli esperti valutano i dati esistenti prima che il produttore possa presentare tutti i documenti altrimenti rilevanti per l'approvazione. Inoltre, controllano continuamente tutti i nuovi risultati ottenuti da ulteriori studi.
Ad esempio, l'EMA ha applicato il processo di revisione continua all'approvazione condizionale del farmaco virale remdesivir durante la pandemia di coronavirus. Nell'ambito del processo di approvazione dei vaccini corona, gli esperti hanno anche verificato i risultati già disponibili e poi ottenuti durante gli studi di fase III in corso.
Medicinali per bambini
I nuovi farmaci di solito passano attraverso diversi studi prima di poter entrare nel mercato. Per molto tempo, tuttavia, un gruppo di pazienti ha ricevuto meno attenzione nella ricerca: bambini e adolescenti. Per il trattamento dei minori, il dosaggio di un farmaco che è stato testato negli adulti è stato spesso semplicemente ridotto.
Dal 2007, tuttavia, ogni nuovo farmaco nell'UE deve essere testato sui minori negli studi di fase II e III se deve essere utilizzato successivamente in questa fascia di età. I test su bambini o adolescenti spesso vengono avviati solo dopo che gli studi di fase II sugli adulti sono stati completati con successo. Un gruppo separato di esperti dell'Agenzia europea per i medicinali EMA, il comitato pediatrico, decide sui dettagli.
I test di ammissione sui minori hanno senso perché i corpi di bambini e adolescenti spesso reagiscono in modo diverso a un farmaco rispetto a quello degli adulti. L'efficacia e la tollerabilità possono quindi essere diverse. Il dosaggio deve quindi essere generalmente aggiustato per i minori. In molti casi, è necessaria una diversa forma di somministrazione per i farmaci per i bambini, come gocce o polvere invece delle compresse grandi che ricevono i pazienti adulti.
Erbe medicinali
Quando si sviluppano nuovi medicinali vegetali (agenti fitoterapici), la prova dell'efficacia, come prescritto sotto forma di studi clinici, è difficile:
Mentre i farmaci chimici di solito non contengono più di una o due sostanze pure, ogni pianta produce una miscela di sostanze attive. Il più delle volte, questo mix varia anche nelle diverse parti della pianta. Ad esempio, l'erba di ortica può influenzare i reni, mentre la radice di ortica può influenzare il metabolismo ormonale della prostata. Inoltre, queste miscele di principi attivi variano notevolmente a seconda dell'origine e della preparazione della pianta, il che influenza anche l'efficacia.
Nel 1978 fu costituito un gruppo di esperti, la cosiddetta Commissione E, per chiarire tali questioni. Questi contengono le informazioni note all'epoca sulla composizione, sugli effetti e sui possibili effetti collaterali delle varie piante medicinali.
Poiché le monografie della Commissione E non sono state aggiornate dal 1994, vengono invece utilizzate le monografie del "Committee on Herbal Medicinal Products" (HMPC). Questo è il comitato dell'Agenzia europea per i medicinali responsabile dei medicinali a base di erbe. Si occupa della valutazione scientifica di tali farmaci.
Occorre distinguere tra fitoterapici tradizionali e fitoterapici moderni: al posto dell'approvazione è necessaria la registrazione. Maggiori informazioni su questo nella prossima sezione.
Registrazione invece di ammissione
I medicinali vegetali tradizionali nonché i preparati omeopatici sono esentati dall'obbligo di autorizzazione come medicinali per "terapie speciali". Occorre invece registrarsi:
Per questo - come per l'approvazione dei medicinali "normali" - deve essere presentata la prova dell'innocuità e dell'appropriata qualità farmaceutica del medicinale omeopatico o vegetale tradizionale.
Nel caso dei medicinali vegetali tradizionali, anche l'effetto farmacologico o l'efficacia devono essere dimostrati in modo plausibile, utilizzando le cosiddette prove tradizionali. Ciò significa che il produttore deve utilizzare le informazioni bibliografiche per dimostrare, tra l'altro, che il medicinale vegetale tradizionale è stato utilizzato a fini medici nell'UE per almeno 30 anni, inclusi almeno 15 anni.
Gli studi clinici per dimostrarne l'efficacia, come prescritto dal classico farmaco di approvazione, non sono però necessari né per le medicine omeopatiche né per le erbe tradizionali affinché un'azienda possa venderle.
A differenza dei farmaci tradizionali nella medicina convenzionale, i rimedi alternativi di solito mancano di ampie prove scientifiche della loro efficacia, soprattutto perché non è richiesto un lungo processo di approvazione dei farmaci.
Tags.: bambino piccolo dieta dormire