Fenilchetonuria
Marian Grosser ha studiato medicina umana a Monaco di Baviera. Inoltre, il dottore, interessato a molte cose, ha osato fare alcune interessanti deviazioni: studiare filosofia e storia dell'arte, lavorare alla radio e, infine, anche per un Netdoctor.
Maggiori informazioni sugli esperti di Tutti i contenuti di sono controllati da giornalisti medici.La fenilchetonuria (PKU) è una malattia ereditaria congenita del metabolismo delle proteine. Previene la degradazione dell'aminoacido fenilalanina. Questo si accumula nel corpo e interrompe lo sviluppo del cervello del bambino. Se non trattata, la fenilchetonuria porta a gravi disabilità intellettive. Con una terapia tempestiva, tuttavia, i pazienti possono condurre una vita normale. Scopri tutto sulla fenilchetonuria qui!
Codici ICD per questa malattia: i codici ICD sono codici riconosciuti a livello internazionale per le diagnosi mediche. Si trovano, ad esempio, nelle lettere dei medici o nei certificati di inabilità al lavoro. E70
Fenilchetonuria: descrizione
La fenilchetonuria (PKU) è una malattia metabolica ereditaria che esiste dalla nascita e interferisce con la scomposizione dell'aminoacido essenziale fenilalanina. Gli amminoacidi sono i mattoni di base delle proteine e quindi componenti metabolici vitali. Alcuni di loro possono entrare nel corpo solo attraverso il cibo, l'organismo non può produrli da solo. Tali amminoacidi sono chiamati essenziali.
Cosa succede con la fenilchetonuria?
Normalmente, gli amminoacidi sono soggetti a un equilibrio tra assorbimento/accumulo e scomposizione, in modo che ce ne sia sempre disponibile quanto il corpo ha bisogno. Nel caso di vari amminoacidi, una carenza o un eccesso può causare danni considerevoli e causare vari sintomi.
Nella cosiddetta PKU classica, l'effetto della fenilalanina idrossilasi (PAH) è limitato o addirittura del tutto assente. A causa della carenza di PAH, la fenilalanina si accumula sempre più nel corpo. Una concentrazione troppo elevata di fenilalanina interrompe considerevolmente lo sviluppo del cervello e porta a disabilità intellettive nei giovani pazienti in una fase iniziale.
Poiché la normale degradazione della fenilalanina non è possibile con la malattia, si formano altri prodotti di degradazione, i cosiddetti fenilchetoni. Sono escreti nelle urine e sono responsabili del nome della malattia.
Fenilchetonuria atipica
Anche con forme atipiche di fenilchetonuria, la degradazione della fenilalanina è disturbata. Tuttavia, la causa non è un difetto della PAH. Invece, la funzione di un coenzima, la tetraidrobiopterina (BH4), è limitata. È indirettamente coinvolto nella degradazione della fenilalanina perché la PAH ha bisogno di BH4 per convertire la fenilalanina in tirosina.
Poiché BH4 è importante anche per la produzione delle sostanze messaggere dopamina e serotonina, la fenilchetonuria atipica con deficit di BH4 è solitamente più complicata della forma classica.
Chi colpisce la fenilchetonuria?
La PKU è una delle malattie metaboliche congenite più comuni. Si stima che circa un neonato su 7.000 nel mondo lo svilupperà, senza differenze tra ragazze e ragazzi. Poiché si tratta di una malattia ereditaria, spesso sono colpiti diversi membri di una famiglia.
Fenichetonuria: sintomi
All'inizio, i bambini con fenilchetonuria non mostrano alcun sintomo della malattia. I primi problemi non si manifestano fino al quarto-sesto mese di vita, se la malattia non è stata ancora riconosciuta e curata. Soprattutto, la maturazione cerebrale disturbata causa enormi complicazioni nel tempo. I sintomi della PKU non trattata includono:
- un forte deficit mentale. Il danno cerebrale progredisce durante la pubertà e poi ristagna. I bambini colpiti sono quindi di solito gravemente handicappati mentali.
- Convulsioni (convulsioni) (epilessia). A causa del danno, le cellule nervose del cervello sono particolarmente sensibili e sovraeccitabili. Il risultato sono frequenti crisi epilettiche.
- disabilità motorie. Non solo le cellule cerebrali, ma anche i muscoli del paziente possono essere sovraeccitati. Ecco perché spesso diventa teso (spasticità), che porta a vari disturbi del movimento.
- Disturbi del comportamento. Alcuni bambini con fenilchetonuria sono iperattivi e insolitamente aggressivi, e anche gli scoppi di rabbia sono più comuni.
- una piccola testa (microcefalia). Poiché il cervello del paziente non si sviluppa correttamente, anche la crescita della testa rimane indietro. La piccola circonferenza della testa rispetto ai loro coetanei è particolarmente evidente nei bambini più grandi.
- un odore notevole. La PKU produce alcuni prodotti di degradazione della fenilalanina che hanno un odore simile agli escrementi di topo. Queste sostanze vengono escrete principalmente nelle urine, ma anche in parte attraverso la pelle.
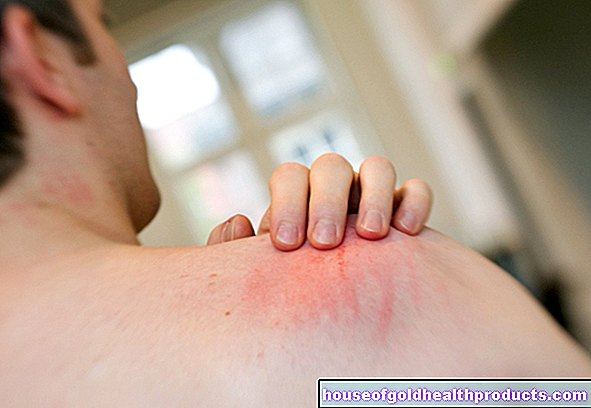
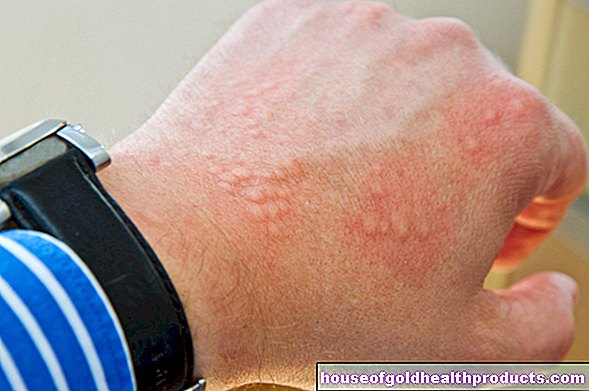
- cambiamenti della pelle simili all'eczema
Poiché anche la produzione del pigmento melanina è disturbata nella fenilchetonuria, molti malati hanno una pelle molto chiara, sensibile al sole e capelli biondo-bianchi. Anche l'iride degli occhi è da blu chiaro a trasparente e lascia trasparire il fondo rossastro.
I sintomi della PKU variano in gravità da persona a persona. La ragione principale di ciò è che l'attività della fenilalanina idrossilasi (PAH) è limitata in modo diverso in ciascun paziente. Alcuni hanno ancora una certa attività residua, per cui nell'organismo si accumula meno fenilalanina. Altri non mostrano alcuna attività enzimatica - la malattia progredisce di conseguenza più velocemente e più seriamente.
Fenilchetonuria: cause e fattori di rischio
La fenilchetonuria è una malattia ereditaria. Sono ormai note numerose mutazioni genetiche che portano a un difetto della PAH. Il tipo di mutazione determina la misura in cui è limitata la degradazione della fenilalanina.
La PKU è ereditata in modo recessivo, il che significa che una persona può essere portatrice di un gene alterato senza sviluppare la malattia. Allo stesso modo, le persone con fenilchetonuria possono generare figli sani.
Solo se entrambi i genitori hanno mutazioni nel loro corredo genetico c'è una certa probabilità che i loro figli sviluppino fenilchetonuria. Se i genitori sono entrambi non solo portatori dei geni, ma hanno anche la PKU, anche tutti i bambini insieme la svilupperanno.
Fenilchetonuria: esami e diagnosi
Poiché le gravi conseguenze della fenilchetonuria possono essere prevenute iniziando il trattamento in tempo utile, è particolarmente importante scoprire la malattia il prima possibile. In Germania, i bambini vengono esaminati per varie malattie congenite, inclusa la PKU, nell'ambito di un esame generale (screening neonatale) il terzo giorno dopo la nascita.
Spettrometria di massa tandem
Molti disordini metabolici congeniti vengono ora diagnosticati con l'aiuto della cosiddetta spettrometria di massa tandem. Consente di esaminare il sangue del neonato in modo rapido e semplice. Oltre alla fenilchetonuria, i medici possono rilevare più di 20 altre malattie in pochi minuti.
prova di Guthrie
Il test di Guthrie, dal nome del suo inventore, consente anche di diagnosticare la PKU. Per fare ciò, una piccola quantità di sangue viene prelevata dal tallone del bambino e applicata a un pezzo di carta da filtro. In laboratorio è quindi possibile determinare se la concentrazione di fenilalanina è aumentata.
Il test di Guthrie è stato introdotto negli anni '60 ed è stato a lungo il metodo standard per la diagnosi della fenilchetonuria. Tuttavia, presenta degli svantaggi rispetto alla spettrometria di massa tandem. Il test di Guthrie fornisce un risultato solo dopo cinque giorni, che è anche soggetto a errori. Ad esempio, fattori come la dieta del bambino o l'eventuale terapia antibiotica falsificano i risultati. In alcuni paesi viene ancora utilizzato il test di Guthrie, in Germania di solito non viene più utilizzato.
Altri test
Se lo screening neonatale rivela un sospetto di fenilchetonuria, segue un ulteriore esame per la conferma. Questo può essere utilizzato anche per determinare l'esatta concentrazione di fenilalanina nel sangue.
Infine, è importante distinguere se si tratta di una PKU tipica (classica) o atipica. Sono disponibili anche test speciali per questo, come lo stress test della tetraidrobiopterina. La distinzione è importante perché la fenilchetonuria atipica viene trattata in modo diverso rispetto alla forma classica.
Esame del liquido amniotico
La fenilchetonuria può essere diagnosticata già durante la gravidanza (diagnosi prenatale). Per fare ciò, viene prelevata una piccola quantità di liquido amniotico dal sacco amniotico della madre e vengono esaminate le cellule del nascituro in esso contenute. Eventuali difetti genetici che causano la PKU possono essere identificati in questo modo.
Con lo screening neonatale, tuttavia, la fenilchetonuria viene solitamente scoperta abbastanza presto da essere trattata. Poiché un test del liquido amniotico è sempre associato a un certo rischio, il suo uso per diagnosticare la PKU di solito non è utile.
Fenilchetonuria: trattamento
C'è solo un modo per contrastare l'eccesso di fenilalanina nella PKU: i bambini affetti devono seguire una dieta speciale per assumere meno fenilalanina possibile con il cibo. Hanno anche bisogno di alcuni integratori alimentari per sostituire le sostanze che si formerebbero dalla fenilalanina nei bambini sani.
La terapia deve iniziare prima che compaiano i primi sintomi di un disturbo dello sviluppo, cioè entro i primi due mesi di vita. Il danno cerebrale che si è già verificato non può essere invertito.
Terapia nutrizionale con la dieta PKU
Tutte le proteine naturali sono costituite da circa il cinque percento dell'amminoacido fenilalanina. La maggior parte degli alimenti ne contiene molto di più di quanto il corpo abbia bisogno. Questo non è un problema per una persona sana perché si scompone ed espelle le quantità in eccesso di fenilalanina.
I pazienti con fenilchetonuria, d'altra parte, devono fare a meno delle proteine naturali per la maggior parte. Prodotti speciali di fabbricazione industriale sostituiscono i componenti alimentari mancanti. Da un lato si vuole prevenire un eccesso di fenilalanina, dall'altro al paziente vanno fornite sostanze che altrimenti sarebbero carenti, come la tirosina, che è formata dalla fenilalanina.
Tuttavia, l'obiettivo della dieta PKU non è fermare completamente l'assorbimento della fenilalanina. Perché l'organismo ha bisogno di una certa quantità di amminoacido per importanti processi metabolici. Raggiungere la giusta concentrazione rappresenta una grande sfida per medici e pazienti e richiede molta disciplina.
La dieta va quindi rispettata per tutta la vita, ma in modo particolarmente rigoroso fino all'età di sei anni. Perché il cervello si sviluppa molto fortemente fino a questa età ed è quindi particolarmente suscettibile ai danni. Nell'età adulta, alte concentrazioni di fenilalanina non causano danni cerebrali come nei bambini. Tuttavia, possono essere l'innesco per altri disturbi neurologici come scarsa concentrazione o reazioni rallentate.
Il trattamento dietetico per la fenilchetonuria dovrebbe iniziare presso un centro specializzato in malattie metaboliche. Perché non è possibile in ogni clinica istruire i genitori sulla dieta. È meglio farlo con l'aiuto di una consulenza dietetica, che mostra come seguire la dieta e monitorare regolarmente i livelli di fenilalanina nel sangue.
Trattamento della fenilchetonuria atipica
Nelle forme atipiche di PKU, il coenzima BH4 mancante e alcune sostanze messaggere come la dopamina e la serotonina vengono sostituite artificialmente. In alcuni casi, il paziente deve anche seguire una dieta a basso contenuto di fenilalanina.
Fenilchetonuria durante la gravidanza
Se sei incinta e soffri di fenilchetonuria, dovresti considerare quanto segue:
- Attieniti alla tua dieta in modo particolarmente rigoroso!
- Fai controllare i valori ematici della fenilalanina a intervalli frequenti. Perché le concentrazioni di fenilalanina nel nascituro sono circa il doppio di quelle della donna incinta. Oltre al cervello, possono danneggiare anche il cuore e gli occhi del nascituro. Malformazioni dello scheletro del bambino possono anche derivare da alti livelli di fenilalanina durante la gravidanza.
- Al fine di escludere danni cerebrali al bambino all'inizio della gravidanza, le donne con fenilchetonuria devono pianificare attentamente la gravidanza e prendere precauzioni fin dall'inizio.
- Nelle donne in gravidanza con fenilchetonuria, una dieta non rigorosamente rispettata può portare a un aborto spontaneo (aborto spontaneo).
- In ogni caso, le donne in gravidanza con fenilchetonuria dovrebbero chiedere consiglio e guida a un medico.
Fenilchetonuria: decorso della malattia e prognosi
Se la fenilchetonuria viene diagnosticata precocemente, se possibile nel neonato, e se viene seguita la dieta speciale per PKU, la prognosi è generalmente buona. I bambini si sviluppano spiritualmente e hanno un'aspettativa di vita media.
Se non trattato, tuttavia, il danno cerebrale porta a gravi disturbi dello sviluppo mentale che non possono essere corretti in seguito. Le persone colpite hanno un'aspettativa di vita normale, ma il loro quoziente di intelligenza è quasi sempre molto al di sotto della norma.
La rara forma atipica di fenilchetonuria, in cui è presente un deficit di BH4, è un caso particolare. Questa variante della fenilchetonuria può portare a un danno neurologico progressivo con forti crampi nonostante la dieta.
Tags.: piante velenose di fungo velenoso nutrimento menopausa










.jpg)