Talassemia
Tanja Unterberger ha studiato giornalismo e scienze della comunicazione a Vienna. Nel 2015 ha iniziato il suo lavoro come redattrice medica presso in Austria. Oltre a scrivere testi specialistici, articoli di riviste e notizie, il giornalista ha anche esperienza nel podcasting e nella produzione di video.
Maggiori informazioni sugli esperti di Tutti i contenuti di sono controllati da giornalisti medici.La talassemia, o anemia mediterranea, è una malattia genetica dei globuli rossi. Un gene difettoso fa sì che il corpo produca troppo poco pigmento rosso del sangue (emoglobina) o si decomponga troppo rapidamente. A seconda della posizione del difetto genetico, viene fatta una distinzione tra alfa e beta talassemia. Entrambe le forme portano all'anemia (anemia). Il medico di solito li tratta con farmaci e trasfusioni di sangue. Leggi di più su cause, sintomi, diagnosi e trattamento qui!
Codici ICD per questa malattia: i codici ICD sono codici riconosciuti a livello internazionale per le diagnosi mediche. Si trovano, ad esempio, nelle lettere dei medici o nei certificati di inabilità al lavoro. D57D56

Breve panoramica
- Descrizione: Malattia geneticamente determinata dei globuli rossi (eritrociti) che porta all'anemia.
- Diagnosi: Il medico diagnostica la talassemia attraverso uno speciale esame del sangue e un'analisi del materiale genetico (analisi del DNA).
- Cause: un difetto genetico ereditario che induce il corpo a produrre troppo poco o nessun pigmento rosso del sangue (emoglobina).
- Trattamento: il trattamento consiste in farmaci e trasfusioni di sangue, in alcuni casi è necessario un trapianto di midollo osseo.
- Sintomi: tra le altre cose, anemia, stanchezza, ingrossamento del fegato e della milza, disturbi della crescita, alterazioni ossee, osteoporosi; forme lievi di talassemia spesso non hanno sintomi.
- Prognosi: le talassemie hanno diversi gradi di gravità. Prima vengono trattati i sintomi, migliore è la prognosi. Una cura è attualmente possibile solo con un trapianto di cellule staminali o una terapia genica.
Cos'è la talassemia?
Le talassemie (anche anemia mediterranea) sono un gruppo di malattie genetiche in cui la formazione dei globuli rossi (eritrociti) è disturbata da un difetto genetico. Di conseguenza, il corpo produce troppo poco o nessun pigmento rosso del sangue (emoglobina) o viene scomposto troppo rapidamente.
A causa dell'emoglobina modificata, le cellule del sangue sono generalmente più piccole del normale e hanno una durata di vita più breve, che porta all'anemia nel tempo, accompagnata da sintomi tipici come stanchezza e battito cardiaco accelerato.
La talassemia è una delle malattie del sangue più comuni nei bambini e negli adolescenti. Appartengono anche al gruppo delle emoglobinopatie (disturbi dell'emoglobina). La talassemia può essere trasmessa di generazione in generazione. Poiché la talassemia è particolarmente diffusa nella regione mediterranea, è anche conosciuta come "anemia mediterranea".
Come si sviluppa la talassemia?
L'emoglobina è una proteina presente nei globuli rossi e prodotta nel midollo osseo. Consente ai globuli rossi di trasportare l'ossigeno vitale dai polmoni a tutte le aree del corpo.
L'emoglobina è solitamente composta da quattro catene proteiche, due delle quali sono uguali: due catene alfa e due catene beta. L'emoglobina contiene anche ferro, che lega l'ossigeno. Nella talassemia, il corpo forma catene proteiche no, troppo poche o modificate (catene alfa o beta) a causa di un gene alterato (mutazione).
Ciò significa che si possono formare meno molecole funzionali di emoglobina. I globuli rossi si restringono e si rompono sempre di più. Ciò porta all'anemia, poiché il corpo non è più adeguatamente rifornito di ossigeno a causa della mancanza di globuli rossi.
A seconda di quale catena proteica (catena alfa o beta) è interessata, viene fatta una distinzione tra α- (alfa) -talassemia e β- (beta) -talassemia.
Alfa talassemia
La forma meno comune è l'α-talassemia. In questa forma, il corpo forma troppo poche o nessuna catena alfa.
beta talassemia
La beta talassemia è la forma più comune di talassemia. In questa forma, il corpo produce catene di emoglobina beta troppo piccole o assenti.
C'è anche delta e gamma talassemia, ma entrambi sono molto rari e generalmente lievi.
Come viene diagnosticata la talassemia?
Poiché i sintomi della talassemia compaiono spesso durante l'infanzia, il primo punto di contatto è solitamente il pediatra. Se necessario e per ulteriori esami, ti indirizzerà ad uno specialista in medicina interna (ematologo).
Storia famigliare
Poiché la talassemia è una malattia ereditaria, la storia familiare di solito fornisce al medico gli indizi iniziali. Ad esempio, il medico chiede se ci sono portatori noti della malattia in famiglia.
analisi del sangue
Per confermare la diagnosi, il medico eseguirà un esame del sangue che include l'elettroforesi dell'emoglobina (elettroforesi dell'Hb).
Elettroforesi dell'emoglobina
Con l'elettroforesi dell'emoglobina, il medico scioglie l'emoglobina dal sangue del paziente in un liquido e la applica su uno speciale materiale di supporto (ad es. di carta o gelatina). Quindi applica una tensione.
A seconda di come è composta l'emoglobina, si sposta a distanze diverse in un dato tempo. In base alla distanza percorsa, il medico valuta se l'emoglobina è normale o difettosa.
Al microscopio, il medico di solito riconosce le cellule dalla forma caratteristica nel sangue della persona. Questi sono globuli rossi relativamente pallidi con un deposito di emoglobina di colore più scuro al centro. Le cellule del sangue sono piccole e hanno una scarsa emoglobina.
Diagnosi nei bambini
I medici raccomandano che i neonati che hanno avuto la talassemia in famiglia abbiano un esame del sangue (ad esempio, per lo screening neonatale). Per fare ciò, il medico preleva del sangue (ad esempio dal cordone ombelicale) subito dopo il parto e lo esamina alla ricerca di cambiamenti genetici. In questo modo è possibile riconoscere precocemente la malattia e curarla tempestivamente.
Diagnosi in gravidanza
Nei nascituri, l'esame per la talassemia (diagnosi prenatale) viene effettuato utilizzando un campione di tessuto dalla placenta.
I medici raccomandano ai genitori in attesa che sanno di essere portatori di talassemia di eseguire questa diagnosi prenatale nelle prime dodici settimane di gravidanza.
La talassemia è ereditaria?
La talassemia è ereditaria. Ciò significa che le persone con talassemia ereditano il difetto genetico dai loro genitori e nascono con la malattia.
Ereditarietà: alfa talassemia
L'alfa talassemia ha diversi gradi di gravità. Viene fatta una distinzione tra alfa talassemia minima, minore, intermedia e maggiore. L'espressione dipende dal numero di geni patologici che i genitori trasmettono al bambino. Ci sono quattro geni per la catena alfa, due della madre e due del padre.
Un totale di quattro geni sono coinvolti nella formazione delle catene alfa. Nell'alfa talassemia major, tutti i geni che il bambino ha ereditato dai genitori sono difettosi. È la forma più grave di alfa talassemia. I bambini con questa forma raramente sono in grado di sopravvivere e di solito muoiono prima della nascita o pochi giorni dopo. Solo in pochi casi è possibile mantenere in vita i bambini con l'aiuto di un trapianto di cellule staminali.
Le persone con alfa talassemia minor o intermedia hanno ereditato due o tre geni difettosi dai loro genitori. Sono tra le forme più lievi di alfa talassemia e di solito causano sintomi moderati, lievi o assenti nelle persone colpite.
Se un solo genitore eredita un gene difettoso dal proprio figlio, si parla di alfa talassemia minima. Le persone colpite di solito non hanno sintomi e vivono senza problemi.
Ereditarietà: beta talassemia
La beta talassemia ha anche diversi gradi di gravità. Si dividono in minori, intermedi e maggiori.
La talassemia major (detta anche anemia di Cooley) è la forma più grave. Si verifica quando entrambi i genitori trasmettono il difetto genetico al bambino.
Con la beta-talassemia major, è necessario scambiare regolarmente il sangue della persona interessata (circa ogni tre-quarta settimana) mediante infusioni. Se non trattate, le persone colpite di solito sviluppano gravi deformazioni ossee. I bambini affetti sono anche molto più suscettibili alle infezioni, sono deboli e di solito non si sviluppano in base alla loro età.
La beta talassemia intermedia è una forma intermedia. Le persone con questa forma hanno anche ereditato i geni difettosi da entrambi i genitori. I loro sintomi di solito non sono così pronunciati come nella forma principale. Hai solo bisogno di trasfusioni di sangue occasionalmente.
Nella beta talassemia minor, le persone colpite hanno ereditato solo un gene difettoso della catena dell'emoglobina beta da un genitore. L'altro genitore ha trasmesso al bambino un gene funzionante.
Le persone con beta-talassemia minor di solito non hanno sintomi di anemia lievi. Di norma, non richiedono trattamento. Tuttavia, in quanto portatori di geni, sono in grado di trasmettere la talassemia ai loro figli.
Se entrambi i genitori hanno una beta talassemia minore, c'è una probabilità del 25% che il bambino erediti entrambe le mutazioni e sviluppi una talassemia maggiore o intermedia. C'è una probabilità del 50 percento che il bambino erediti una forma minore e una probabilità del 25 percento che il bambino sia sano.
Per ottenere la beta talassemia, entrambi i genitori devono trasmettere i loro geni difettosi al bambino. Un portatore della malattia ha un solo gene difettoso, l'altro gene è funzionale. Sebbene non si ammalino, possono trasmettere il gene ai loro figli.
Dove si verifica la talassemia?
La talassemia è anche conosciuta colloquialmente come anemia mediterranea, poiché è diffusa nei paesi mediterranei come l'Italia e la Grecia. Ma si verifica anche nel Vicino e Medio Oriente e in alcune parti dell'Africa e dell'Asia.Nell'Europa centrale, la talassemia si è diffusa attraverso i legami commerciali globali e la migrazione.
Dagli anni '60, la talassemia si è verificata anche nell'Europa centrale e settentrionale (ad esempio Germania, Austria, Francia, Inghilterra, Paesi Bassi, Belgio e Scandinavia). Nel frattempo, la talassemia è una delle malattie del sangue più comuni nei bambini e negli adolescenti.
Quanto è comune la malattia?
Si stima che circa 500-600 persone in Germania vivano con una forma grave di talassemia. Circa 200.000 persone in Germania soffrono di una forma lieve di talassemia. Inoltre, le beta talassemie sono più comuni delle alfa talassemie.
Come si cura la talassemia?
Il medico, di solito uno specialista (ad esempio un ematologo pediatrico), tratta la talassemia a seconda della gravità della malattia e dei sintomi che si verificano. Il trattamento avviene in centri specializzati nella malattia. A seconda che la persona interessata abbia bisogno di una trasfusione di sangue, viene ora fatta una distinzione tra terapia trasfusione-dipendente (TDT) e non trasfusione-dipendente (NTDT) per la talassemia.
Trattamento non dipendente dalla trasfusione
Le forme lievi di talassemia di solito non richiedono trattamento. Tuttavia, in rari casi durante la gravidanza, le donne in gravidanza possono sviluppare una grave anemia a causa di una lieve talassemia. Hanno quindi bisogno di trasfusioni di sangue regolari per proteggere sia la madre che il bambino.
Trattamento dipendente dalla trasfusione
Il medico scambia il sangue di persone dipendenti da trasfusione (ad esempio con talassemia major o intermedia) con trasfusioni di sangue regolari. A seconda della gravità della talassemia, questo di solito si verifica ogni seconda o terza settimana, di solito per tutta la vita.
Le trasfusioni di sangue aggiuntive spesso portano a un eccesso di ferro. In questi casi, il medico prescriverà farmaci che rimuovono il ferro in eccesso dal corpo (i cosiddetti chelanti o chelanti del ferro). Il medico lo inietta sotto la pelle (per via sottocutanea) in ospedale o la persona interessata prende le compresse a casa.
Trapianto di cellule staminali
È possibile curare la talassemia major con un trapianto di cellule staminali. Tuttavia, questo presuppone che ci sia un donatore adatto per la persona interessata.
Terapia genetica
Dal 2019 esiste una terapia genica per la beta talassemia trasfusione-dipendente. La terapia tratta il difetto genetico causale e dà alle persone colpite la possibilità di vivere senza trasfusioni per tutta la vita.
In questo nuovo trattamento, un gene intatto in grado di produrre emoglobina sana viene iniettato nelle cellule staminali del sangue della persona. Per fare ciò, le cellule staminali vengono prima prelevate dal midollo osseo del paziente e trattate con un farmaco. Quindi la persona interessata riceve le proprie cellule staminali (con geni intatti), che ora formano globuli rossi sani e normale pigmento del sangue.
L'Agenzia europea per i medicinali (EMA) ha approvato il trattamento per le persone di età pari o superiore a dodici anni idonee a questa particolare forma di terapia. La terapia genica è attualmente possibile solo in Germania. Pochissimi pazienti sono attualmente trattati con esso.
Dieta e stile di vita
Le persone con talassemia sono in grado di influenzare positivamente il decorso della malattia mangiando la giusta dieta e adottando uno stile di vita appropriato. Si prega di notare quanto segue:
- Mangiare una dieta equilibrata.
- Evita gli alimenti che sono estremamente ricchi di ferro (ad esempio il fegato).
- Ottieni abbastanza calcio dal cibo (ad esempio latte, yogurt, formaggio, foglie di spinaci, broccoli).
- Non bere alcolici.
- Fai esercizio fisico regolare (circa tre o quattro ore di esercizio fisico come camminare velocemente, fare jogging o andare in bicicletta a settimana).
Quali sono i sintomi della talassemia?
La gravità dei sintomi dipende dalla quantità di emoglobina modificata in modo anomalo. Le persone con una forma lieve di talassemia di solito hanno pochi o nessun sintomo.
Se non trattati, tuttavia, i bambini con una forma grave di talassemia mostrano problemi di salute già a quattro o cinque mesi di età.
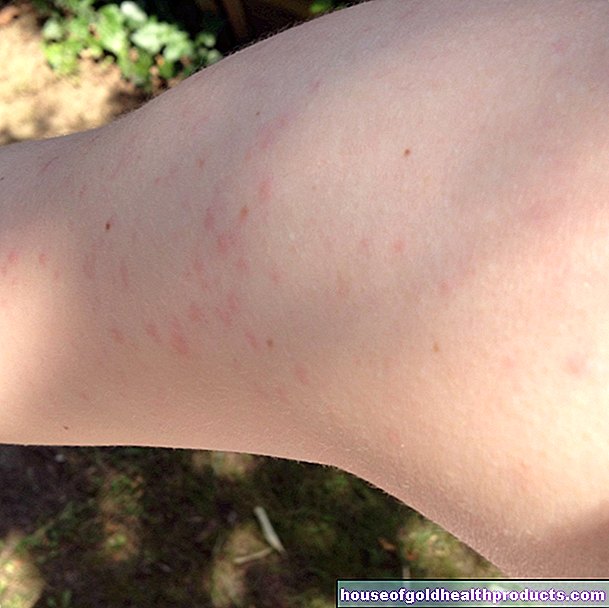
Anemia
Le persone che hanno la talassemia major o intermedia inizialmente mostrano i sintomi tipici dell'anemia:
- Sono pallidi o hanno la pelle giallastra.
- Sei stanco tutto il tempo.
- Hai le vertigini.
- Hai mal di testa.
- Hai un battito cardiaco accelerato.
- Hanno rapidamente affanno durante l'attività fisica.
- Spesso hanno fegato e milza ingrossati.
- Il loro sviluppo è spesso disturbato.
A prima vista, la talassemia assomiglia all'anemia sideropenica. Tuttavia, il livello di ferro è basso in caso di carenza di ferro, mentre è solitamente normale nella talassemia.
In seguito o se il trattamento è inadeguato, le persone colpite sperimentano spesso gravi effetti collaterali. Questi includono, ad esempio:
- Deformazioni ossee
- Infezioni frequenti
- problemi di cuore
- Diabete mellito
- osteoporosi
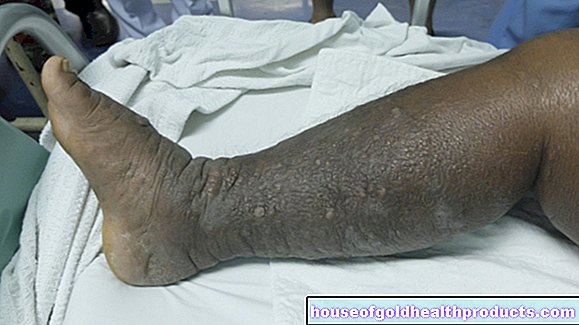
Cambiamenti ossei
Le persone con talassemia major o intermedia spesso mostrano anche una crescita ossea anormale:
- L'osso del cranio si allarga.
- La fronte, la mascella superiore e l'osso zigomatico sono sporgenti.
- Le costole e i corpi vertebrali sono piegati.
- Sei a maggior rischio di fratture.
- Hai dolore alle ossa.
- Ti viene l'osteoporosi.
Ferro in eccesso
Le persone con forme gravi di talassemia spesso immagazzinano troppo ferro nel corpo (sovraccarico di ferro), che non si decompone più da solo. Questo può danneggiare gli organi (emocromatosi secondaria). L'aumento di ferro deriva, tra l'altro, dal sangue in più che la persona interessata riceve regolarmente tramite infusioni.
Inoltre, il corpo considera l'anemia come una carenza di ferro e cerca di compensarla assumendo più ferro dal cibo. Il corpo produce anche cellule del sangue in eccesso. Il fegato e la milza quindi lavorano a tutta velocità per abbatterli di nuovo.
L'eccessiva concentrazione di ferro provoca altri sintomi nelle persone colpite, come:
- Insufficienza cardiaca (insufficienza cardiaca)
- Aritmia cardiaca
- Disfunzione del fegato
- Diabete (diabete mellito)
- Bassa statura
- Pubertà ritardata
- Tiroide ipoattiva (ipotiroidismo)
- Disturbi del metabolismo della vitamina D
Quando dal dottore?
Poiché le forme gravi di talassemia si manifestano spesso nei primi mesi di vita, è importante che i genitori vedano un pediatra il prima possibile ai primi segni (es. pallore).
Come puoi prevenirlo?
I medici consigliano alle persone che sanno di avere il gene per la talassemia di rivolgersi a un centro di consulenza genetica prima di una gravidanza programmata o se vogliono avere figli.
Specialisti formati utilizzano un esame del sangue genetico per determinare i possibili rischi genetici associati alla gravidanza nelle coppie.
Si consiglia inoltre alle persone con una storia familiare nota di talassemia di chiedere consiglio prima di concepire.
Per prevenire infezioni potenzialmente letali, i medici raccomandano anche che i bambini con talassemia e i bambini sani vengano vaccinati secondo l'attuale calendario delle vaccinazioni. In alcuni casi è necessario trattare regolarmente i bambini con talassemia con determinati antibiotici. Questi aiutano a prevenire gravi infezioni batteriche, a cui le persone con talassemia sono particolarmente sensibili.
La misurazione regolare della temperatura corporea aiuta a rilevare le infezioni il prima possibile e a trattarle il più rapidamente possibile. Se hai la febbre superiore a 38,5 gradi Celsius, dovresti consultare immediatamente un medico. È possibile che l'infezione stia causando questo.
È anche particolarmente importante che le persone colpite si facciano controllare regolarmente da un medico. Questo è l'unico modo per monitorare attentamente il decorso della malattia e identificare e trattare le complicanze in una fase iniziale.
La talassemia è curabile?
Una cura completa per la talassemia è attualmente possibile solo con un trapianto di cellule staminali o con la terapia genica. L'aspettativa di vita e la qualità delle persone colpite è aumentata continuamente grazie a nuovi farmaci, migliori cure e una migliore istruzione.
Qual è la prognosi per la talassemia?
Le persone con talassemia lieve (minore alfa o beta talassemia) hanno un'aspettativa di vita normale. Di norma, le persone colpite possono vivere senza disabilità.
Con le forme più gravi, spesso è anche possibile raggiungere un'aspettativa di vita quasi normale con il giusto e tempestivo trattamento (ad es. regolari trasfusioni di sangue). Tuttavia, è quindi necessaria una terapia permanente, continua e intensiva.
A volte il trapianto di cellule staminali può curare la malattia e, in rari casi, la terapia genica.
Tags.: mestruazioni prevenzione rivista