Fibrosi cistica
e Christiane Fux, redattore medico e Martina Feichter, redattrice medica e biologaSophie Matzik è una scrittrice freelance per il team medico di
Maggiori informazioni sugli esperti diChristiane Fux ha studiato giornalismo e psicologia ad Amburgo. L'esperto redattore medico scrive articoli di riviste, notizie e testi fattuali su tutti i possibili argomenti di salute dal 2001. Oltre al suo lavoro per, Christiane Fux è anche attiva nella prosa. Il suo primo romanzo poliziesco è stato pubblicato nel 2012 e scrive, progetta e pubblica anche le sue commedie poliziesche.
Altri post di Christiane FuxMartina Feichter ha studiato biologia con una materia elettiva farmacia a Innsbruck e si è anche immersa nel mondo delle piante medicinali. Da lì non era lontano da altri argomenti medici che la affascinano ancora oggi. Si è formata come giornalista presso l'Axel Springer Academy di Amburgo e lavora per dal 2007 - prima come redattrice e dal 2012 come scrittrice freelance.
Maggiori informazioni sugli esperti di Tutti i contenuti di sono controllati da giornalisti medici.
La fibrosi cistica è una malattia metabolica congenita. Nelle persone colpite, i fluidi corporei come la saliva, il muco bronchiale o le secrezioni pancreatiche sono molto più duri del solito. Ciò causa problemi respiratori e disturbi digestivi, tra le altre cose. Non esiste una cura per la fibrosi cistica. Con una terapia coerente, tuttavia, il decorso della malattia può essere rallentato. Leggi qui i sintomi della fibrosi cistica e come trattarli.
Codici ICD per questa malattia: i codici ICD sono codici riconosciuti a livello internazionale per le diagnosi mediche. Si trovano, ad esempio, nelle lettere dei medici o nei certificati di inabilità al lavoro. E84
Fibrosi cistica: riferimento rapido
- Descrizione: malattia metabolica ereditaria, provoca la formazione di muco denso nei polmoni e in altri organi
- Sintomi: problemi respiratori, tosse secca, suscettibilità alle infezioni dei polmoni, ritardo della crescita, indigestione, grave diarrea, fegato grasso, ridotta fertilità
- Cause: ereditarietà di geni difettosi che influiscono sulla consistenza dei fluidi corporei. Tuttavia, la malattia si manifesta solo se entrambi i genitori trasmettono un gene malato al figlio (ereditarietà autosomica recessiva)
- Diagnostica: esame del sangue per tripsina immunoreattiva (IRT), proteina associata alla pancreatite (PAP), test del sudore, test genetico
- Terapia: ispessenti catarro, broncodilatatori, inalazione, antibiotici per infezioni respiratorie batteriche, cortisone, modulatori CFTR, trapianto di polmone
- Prognosi: incurabile, decorso fortemente dipendente dalla gravità e dal tempo della diagnosi, aspettativa di vita ridotta
Fibrosi cistica: descrizione
La fibrosi cistica (chiamata anche fibrosi cistica) è una malattia metabolica ereditaria. La formazione di vari fluidi corporei è disturbata. Le secrezioni dei polmoni, del pancreas e di altri organi sono più viscose che nelle persone sane.
muco denso
Il muco duro ostruisce, tra le altre cose, i piccoli rami dei bronchi e i dotti degli organi interni. La respirazione e la digestione sono particolarmente colpite. Nel corso della malattia, gli organi possono lavorare sempre meno.
Ecco cosa succede con la fibrosi cistica
Errore nel genoma
La causa della malattia sono difetti nel corredo genetico. La fibrosi cistica non è quindi curabile. Il momento della diagnosi e la gravità dei sintomi possono variare ampiamente da persona a persona. In molti bambini la fibrosi cistica si nota fin dalla nascita, in altri casi viene riconosciuta solo successivamente.
Fibrosi cistica: sintomi
I sintomi della fibrosi cistica possono variare notevolmente da un paziente all'altro. La malattia colpisce la funzione di vari organi, ma soprattutto i polmoni e l'apparato digerente.
Riconosci i primi segnali
Quando compaiono i primi sintomi nella fibrosi cistica varia da persona a persona. Nella maggior parte dei casi, i sintomi della fibrosi cistica compaiono entro il primo anno di vita. Ciò significa che la malattia di solito può essere diagnosticata precocemente e la terapia può essere avviata rapidamente. Tuttavia, alcuni pazienti hanno sintomi chiari solo nell'adolescenza. Inoltre, non tutte le persone colpite mostrano l'intera gamma di possibili sintomi. Anche la gravità dei sintomi varia.
Fluidi corporei alterati
Nella fibrosi cistica, la formazione dei cosiddetti canali ionici cloruro nelle membrane cellulari è disturbata. Questo cambia la composizione dei fluidi corporei. Il modo più semplice per determinare questo cambiamento nel sudore delle persone colpite. Il loro sudore è più salato che nelle persone sane. I sali di sodio e cloruro, che fanno parte dei cosiddetti elettroliti, si accumulano nel sudore. Chi soffre di fibrosi cistica perde più sali corporei attraverso la sudorazione.
Sintomi multipli di fibrosi cistica
La malattia colpisce un certo numero di sistemi di organi. I primi sintomi della fibrosi cistica compaiono spesso nei polmoni e nel tratto digestivo. Ulteriori lamentele possono sorgere nel corso della vita. I sintomi possono essere trattati bene attraverso una terapia mirata. Tuttavia, i sintomi possono anche essere minacciosi. È particolarmente pericoloso quando i bronchi sono bloccati dal muco denso. Quindi, in casi estremi, i pazienti possono soffocare.
-
"Lo sport può alleviare i sintomi"
Tre domande per
Suat Evcumen,
Specialista in medicina interna e pneumologia -
1
Perché la fibrosi cistica viene riconosciuta così tardi in alcune persone?
Suat EvcumenLa fibrosi cistica è una malattia genetica. A seconda della gravità delle mutazioni, ci sono anche corsi lievi che diventano evidenti solo in ritardo. Fondamentalmente, alcuni canali del cloro non funzionano correttamente, motivo per cui le secrezioni corporee del paziente si addensano. Ciò colpisce diversi sistemi di organi, come le vie respiratorie o la digestione. Ecco perché si parla di una malattia multisistemica.
-
2
Consiglia lo screening neonatale?
Suat EvcumenLa diagnosi precoce è estremamente importante e può aiutare a influenzare positivamente il decorso della malattia attraverso una terapia precoce. Questi includono, ad esempio, la somministrazione di enzimi digestivi, la terapia inalatoria e il supporto fisioterapico precoce. Per questo motivo lo screening neonatale è stato effettuato in Germania per un certo numero di anni.
-
3
Cosa significa la diagnosi?
Suat EvcumenL'aspettativa di vita dei pazienti con fibrosi cistica è aumentata costantemente negli ultimi due decenni. Lo dobbiamo al rilevamento tempestivo e all'inizio precoce delle terapie personalizzate. La maggior parte cresce con la malattia e "impara" ad affrontarla. Continuiamo a osservare che l'esercizio aiuta e allevia i sintomi. Inoltre, cerca un aiuto psicologico.
-
Suat Evcumen,
Specialista in medicina interna e pneumologiaDa marzo 2019 Suat Evcümen è il medico senior in pneumologia presso la Clinica Paracelsus a Silbersee.

Fibrosi cistica: sintomi dei polmoni
Difficoltà respiratorie e tosse secca
Nella maggior parte dei casi di fibrosi cistica, i sintomi polmonari non compaiono fino ai bambini leggermente più grandi. I neonati di solito non hanno problemi respiratori. Nei bambini leggermente più grandi, i sintomi della fibrosi cistica spesso assumono la forma di tosse irritabile cronica simile alla pertosse. Il muco nelle loro vie aeree è aumentato, denso e denso. Ciò ostruisce il flusso d'aria nei polmoni. Nel tempo, si sviluppa difficoltà di respirazione.
Infezioni frequenti
L'aumento dell'accumulo di muco nei polmoni rende più facile l'insediamento dei batteri e la causa di un'infezione. Le polmoniti ricorrenti o le infezioni bronchiali sono causate principalmente da batteri come gli stafilococchi e le specie Pseudomonas. L'equilibrio alterato del sale nei polmoni ostacola anche le difese dell'organismo. Può verificarsi anche sanguinamento polmonare. Un tipico segno di questo è la tosse con muco misto a sangue.
Sebbene i polmoni siano già danneggiati dalla prima infanzia, i primi sintomi della fibrosi cistica nelle vie aeree spesso compaiono solo in età scolare o anche più tardi. I sintomi a volte sono evidenti solo quando ampie parti del tessuto polmonare sono già state distrutte o le vie aeree sono gravemente ristrette.
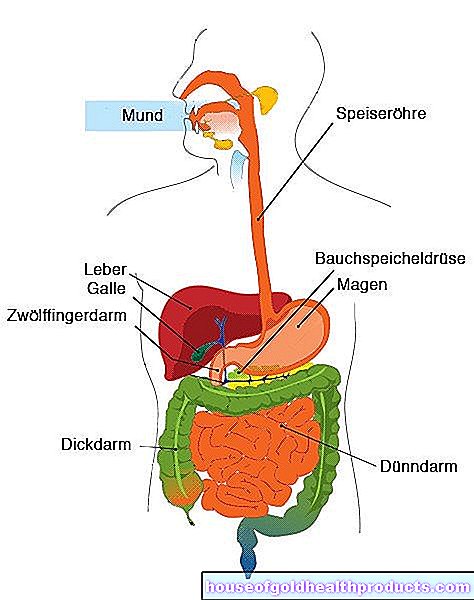
Fibrosi cistica: sintomi del pancreas
Nei pazienti con fibrosi cistica, il pancreas si infiamma spesso: secerne una secrezione che contiene, tra l'altro, enzimi per la digestione dei grassi e dei carboidrati (funzione pancreatica esocrina). Nelle persone con fibrosi cistica, la secrezione si accumula a causa della sua viscosità e provoca l'infiammazione del pancreas (pancreatite).
Man mano che il processo progredisce, il tessuto pancreatico si indurisce e diventa cicatriziale. I medici parlano di fibrosi. La fibrosi distrugge gradualmente il pancreas.
Oltre alla sua secrezione digestiva, il pancreas produce anche l'ormone insulina (funzione pancreatica endocrina) in cellule speciali - le cellule beta. Tra le altre cose, è necessario che il corpo utilizzi lo zucchero. Nei pazienti con fibrosi cistica, la secrezione digestiva viscosa nei dotti pancreatici distrugge gradualmente le cellule beta - la produzione di insulina è compromessa e si sviluppa una carenza di insulina (specialmente nei pazienti di età superiore ai 10 anni). Di conseguenza, può svilupparsi una forma di diabete (diabete mellito): diabete di tipo 3.
Fibrosi cistica: sintomi della bile
Il pancreas e la cistifellea condividono un dotto comune nell'intestino. Pertanto, l'arretrato delle secrezioni pancreatiche può anche causare l'infiammazione della cistifellea. Spesso si formano anche calcoli biliari, che possono bloccare completamente il drenaggio della bile dalla cistifellea.
Fibrosi cistica: sintomi dell'apparato digerente
Oltre ai disturbi dei polmoni, la fibrosi cistica causa molto spesso sintomi nel tratto digestivo. Ad esempio, la mancanza di bile influisce sulla digestione dei grassi. Di conseguenza, i pazienti hanno spesso difficoltà a tollerare i cibi grassi. La maggior parte del cibo ingerito viene escreto non digerito. Sgabelli molto voluminosi e morbidi sono tipici.
Diarrea e crescita stentata
I bambini affetti, compresi i neonati, spesso soffrono di grave diarrea. Anche se bevono e mangiano a sufficienza, difficilmente aumentano di peso. Crescita stentata e malnutrizione sono quindi ulteriori conseguenze classiche della malattia.
Tale indigestione può verificarsi anche con molte altre malattie. Solo in combinazione con problemi respiratori sono un'indicazione di fibrosi cistica, che dovresti assolutamente indagare.
prolasso anale
Man mano che progredisce, la fibrosi cistica può portare a varie complicazioni nel tratto digestivo. Il più comune è quello che è noto come prolasso anale. Parte della membrana mucosa dell'ano fuoriesce dall'ano. Un tale incidente deve essere operato il prima possibile.
Ostruzione intestinale
Frequentemente si verifica anche un'invaginazione dell'intestino (intussuscezione) o un'ostruzione intestinale (ileo). Entrambe le complicazioni sono accompagnate da forti dolori addominali e significativi problemi digestivi. Il dolore di solito si manifesta in periodi, soprattutto dopo aver mangiato. Un'ostruzione intestinale è fatale se non trattata. Il dolore addominale spasmodico e acuto dovrebbe quindi essere sempre chiarito da un medico.
Fibrosi cistica: sintomi del fegato
fegato grasso
Il fegato soffre anche dell'accumulo di bile. Il fegato grasso si sviluppa in molti pazienti con il progredire della malattia. Questo può essere accompagnato da affaticamento, perdita di appetito, sensazione di pienezza e flatulenza e, in rari casi, sensazioni di pressione o leggero dolore nella parte superiore dell'addome.
restringere il fegato
In rari casi, i pazienti con fibrosi cistica sviluppano il fegato rimpicciolito (cirrosi epatica). Questa grave disfunzione epatica diventa prima evidente sotto forma di ittero (ittero). Una colorazione giallastra del bianco degli occhi è un segno di ittero. I problemi cardiaci si verificano anche dopo un lungo periodo di tempo e le prestazioni delle persone colpite continuano a diminuire.
Fibrosi cistica: diminuzione della fertilità
Più della metà di tutti i pazienti maschi di fibrosi cistica sono sterili. Nella maggior parte dei casi, le persone colpite producono sperma fertilizzabile nei loro testicoli. Tuttavia, questi non possono passare attraverso i dotti deferenti perché sono bloccati dal muco viscoso.
Le donne con fibrosi cistica sono generalmente meno fertili. In linea di principio, possono concepire e portare in grembo un bambino. Tuttavia, il muco duro si accumula nelle tube di Falloppio, che lo sperma difficilmente può penetrare. La probabilità di gravidanza diminuisce rapidamente, soprattutto quando le persone invecchiano.
Fibrosi cistica: sintomi nei bambini
La fibrosi cistica è una malattia genetica. È quindi sempre presente fin dalla nascita. Ma i classici sintomi della fibrosi cistica non compaiono sempre durante l'infanzia. Tuttavia, ci sono spesso suggerimenti non specifici che dovrebbero essere seguiti. Ciò è particolarmente vero se la famiglia ha già avuto casi di fibrosi cistica.
Sintomi nel tratto digestivo
Un'indicazione di un disturbo metabolico è, ad esempio, uno stomaco gonfio per lungo tempo. I bambini spesso soffrono di diarrea. Nei neonati, un primo movimento intestinale molto ritardato (Kindspech) può essere un'indicazione di fibrosi cistica. In molti casi, si verificano problemi di crescita e acrobazie anche se i bambini mangiano con appetiti famelici. La stitichezza a causa della malattia si verifica solo in rari casi.
Sintomi nel tratto respiratorio
Altri sintomi che possono indicare la fibrosi cistica sono la respirazione rumorosa e una grave irrequietezza. Molti bambini soffrono di infiammazione cronica del seno. Ciò è particolarmente evidente attraverso il dolore al viso che non può essere localizzato con precisione. I polipi nasali sono anche più comuni nei bambini con fibrosi cistica rispetto ai bambini sani.
Se i bambini soffrono di problemi respiratori o di indigestione per lungo tempo, è sempre consigliabile consultare un medico come misura precauzionale. Situazioni pericolose per la vita possono sorgere rapidamente nei bambini perché non possono articolare da soli le loro lamentele o non possono valutarne la gravità.
Fibrosi cistica: cause e fattori di rischio
La fibrosi cistica è causata da un difetto genetico. Il cambiamento patologico è sul settimo cromosoma nel cosiddetto gene CFTR.
Il gene CFTR (gene regolatore transmembrana della fibrosi cistica) contiene le istruzioni per la costruzione di un canale nella membrana cellulare attraverso il quale gli ioni cloruro entrano nella cellula. I canali ionici cloruro difettosi bloccano il trasporto del sale in alcune cellule del corpo nei pazienti con fibrosi cistica.
Le cellule della ghiandola colpite secernono quindi muco duro invece della secrezione altrimenti liquida. Nei polmoni, nei seni paranasali, nel pancreas, nell'intestino, nelle vie biliari e nelle gonadi si formano secrezioni mucose dure ad alto contenuto di sale.
Fibrosi cistica: quanto è a rischio mio figlio?
La fibrosi cistica si manifesta solo quando entrambi i genitori trasmettono al figlio un gene CFTR patologicamente alterato. Questi genitori di solito sono entrambi sani - hanno un gene CFTR sano e uno difettoso.
Le persone con fibrosi cistica sono solo parzialmente fertili. Alcuni pazienti diventano ancora genitori. I padri o le madri malati trasmettono sempre un gene malato, poiché entrambi i geni CFTR trasportano le informazioni sulla fibrosi cistica. Tuttavia, i tuoi figli si ammaleranno solo se hanno ricevuto anche un gene malato dall'altro genitore.
Le coppie che hanno avuto casi di fibrosi cistica nelle loro famiglie dovrebbero cercare una consulenza genetica prima di pianificare una gravidanza.
Ecco come si eredita la fibrosi cistica
Diagnostica preimpianto
I genitori che potrebbero trasmettere la fibrosi cistica al proprio figlio possono usufruire della diagnostica preimpianto. Le cellule uovo vengono prima fecondate artificialmente. Le prime divisioni cellulari avvengono in provetta (in vitro).
Prima che un embrione così creato venga inserito nell'utero della donna, vengono controllate le proprietà genetiche alterate.Vengono quindi impiantati solo gli embrioni che non contengono il gene malato.
Indipendentemente da ciò, si può anche esaminare durante la gravidanza se il bambino svilupperà successivamente la fibrosi cistica.
Fibrosi cistica: esami e diagnosi
A differenza di alcuni anni fa, la maggior parte degli ospedali ora effettua regolarmente lo screening per i neonati. Include, tra le altre cose, un esame per la fibrosi cistica. La partecipazione a questo processo di screening è volontaria e richiede il consenso dei genitori del neonato.
Screening nel sangue e nel sudore
Il sangue viene prelevato dal neonato per lo screening. Il test per la fibrosi cistica ha diverse fasi:
- Esame del sangue: test per l'aumento dei livelli di tripsina immunoreattiva (IRT) e proteina associata alla pancreatite (PAP). Se ci sono anomalie, viene eseguito il test del sudore.
- Test del sudore: le persone con fibrosi cistica hanno un contenuto di sale significativamente più alto nel sudore rispetto alle persone sane. Per il test del sudore della fibrosi cistica, viene misurato il contenuto dei sali di sodio e cloruro nel sudore corporeo. Nei bambini, il sudore viene raccolto sull'avambraccio e poi analizzato. Se sorge un sospetto qui, viene eseguito un test genetico.
Test genetico
Nei pazienti con fibrosi cistica, il cosiddetto gene CFTR, che fornisce le istruzioni per la costruzione di determinati canali ionici, viene modificato. Queste istruzioni sono lunghe, consistono di circa 6500 paia di basi. Un errore può insinuarsi nel codice ovunque - la fibrosi cistica può quindi essere basata su vari cambiamenti genetici (mutazioni). Questi hanno effetti diversi. Pertanto, vengono testate solo le deviazioni più importanti nel gene CFTR.
Se entrambi i geni CFTR, quello del padre e quello della madre, mostrano una mutazione, questa - insieme al test del sudore positivo - conferma la diagnosi di fibrosi cistica.
La storia familiare fornisce indizi
Se non è stato effettuato un adeguato screening neonatale e il sospetto di fibrosi cistica sorge successivamente, il medico di famiglia o un internista è il contatto giusto. In una prima conversazione, registra la storia medica (anamnesi). Se si sospetta la fibrosi cistica, l'obiettivo principale è l'anamnesi familiare (ci sono malattie note della fibrosi cistica in famiglia? Uno dei genitori è noto per essere portatore di un gene CFTR difettoso?).
Indagini
Verrà quindi effettuato un esame fisico. Il medico ascolta i polmoni del paziente e scansiona i suoi organi interni. In questo modo, può già escludere alcune altre malattie associate a sintomi simili alla fibrosi cistica.
Il test del sudore e il test genetico, come descritto sopra, aiutano anche se si sospetta la fibrosi cistica nei bambini più grandi, negli adolescenti o negli adulti.
Una volta che la diagnosi è stata fatta, il medico può utilizzare vari esami per verificare fino a che punto gli organi interni sono compromessi nella loro funzione e se stanno già emergendo complicanze o malattie secondarie. Tali esami vengono talvolta eseguiti di routine per monitorare la progressione della fibrosi cistica.
Ad esempio, gli esami del sangue possono essere utilizzati per determinare se la secrezione di insulina dal pancreas è ridotta e il livello di zucchero nel sangue è aumentato - sintomi del diabete. I valori della funzionalità epatica e la concentrazione di vitamine liposolubili possono essere misurati anche nel sangue.
La concentrazione dell'enzima digestivo elastasi (proviene dal pancreas) e la concentrazione di grasso sono determinate in campioni di feci.
I campioni di tessuto della gola o dell'espettorato del paziente vengono esaminati in laboratorio per le infezioni batteriche.
Anche le procedure di imaging (come i raggi X, la tomografia computerizzata = TC) sono spesso rivelatrici. Possono essere utilizzati per rilevare infezioni polmonari, ad esempio. I polipi nasali possono essere rilevati anche mediante TC.
Le misurazioni della funzionalità polmonare (test di funzionalità polmonare) mostrano il buon funzionamento dei polmoni.
Testare i membri della famiglia
Se la fibrosi cistica si trova in una famiglia, ha senso che anche tutti gli altri membri della famiglia si sottopongano a un esame. La fibrosi cistica può manifestarsi anche in forme lievi. Quindi spesso ci vogliono molti anni prima che la fibrosi cistica diventi evidente con sintomi chiari. Tuttavia, la diagnosi precoce e la terapia della fibrosi cistica sono importanti anche per questi pazienti, al fine di aumentare l'aspettativa di vita.
Fibrosi cistica: trattamento
Non esiste una cura per la fibrosi cistica. I bambini nati con fibrosi cistica soffriranno degli effetti della malattia per il resto della loro vita. Con una combinazione di fisioterapia, farmaci e inalazioni, il decorso della malattia può essere notevolmente rallentato. L'obiettivo principale della terapia della fibrosi cistica è quello di consentire alle persone colpite di condurre una vita il più normale possibile.
Imparare a convivere con la malattia
È particolarmente importante che i bambini imparino ad affrontare la malattia a lungo termine. I bambini con fibrosi cistica dovrebbero imparare il prima possibile cosa significa la malattia e come colpisce il corpo. A tal fine può essere utile un ricovero ospedaliero con unità formative speciali. Bambini e genitori imparano come dovrebbero mangiare, com'è lo sport e come comportarsi al meglio in situazioni critiche. Genitori e bambini sono anche informati sulle misure igieniche (secondo le raccomandazioni del Robert Koch Institute): igiene delle mani regolare ed estesa (lavaggio delle mani con acqua e sapone), nessun mobile imbottito e piante in vaso.
Aiuto per i polmoni
Ci sono varie opzioni per trattare i sintomi della fibrosi cistica. Quale di questi sia utile nei singoli casi dipende dall'età del paziente e dalla gravità dei sintomi.
espettoranti
I pazienti con fibrosi cistica soffrono maggiormente di problemi polmonari. L'inalazione regolare con additivi speciali (agenti mucolitici) scioglie il muco duro e facilita l'espettorazione.
Broncodilatatori
I cosiddetti simpaticomimetici beta-2 allargano i bronchi, il che rende la respirazione ancora più facile.
Medicinali per le infezioni respiratorie
La scarsa ventilazione dei polmoni rende le persone con fibrosi cistica più inclini alle infezioni respiratorie batteriche. Le persone colpite dovrebbero essere trattate con antibiotici in una fase iniziale. Il medico curante prescrive spesso antibiotici da inalare, perché i principi attivi raggiungono direttamente il sito bersaglio (polmoni). In alternativa o in aggiunta, può essere indicata l'assunzione di antibiotici (ad es. sotto forma di compresse).
Nelle infezioni gravi, spesso è necessario somministrare antibiotici in vena (per via endovenosa). Ciò può essere necessario in caso di infezioni croniche da Pseudomonas aeruginosa, ad esempio.
Le infezioni respiratorie fungine possono essere trattate con agenti antimicotici (antimicotici). I farmaci che inibiscono i virus (antivirali) possono spesso aiutare con le infezioni respiratorie virali.
Farmaci antinfiammatori
In molti pazienti, le vie aeree sono frequentemente o cronicamente infiammate. Quindi vengono utilizzati farmaci antinfiammatori, in particolare glucocorticoidi (cortisone). Possono essere inalati, assunti per via orale o somministrati in vena.
Modulatori CFTR
Da alcuni anni sono disponibili i cosiddetti modulatori CFTR per il trattamento della fibrosi cistica. Questi farmaci, assunti a lungo termine, possono migliorare la funzione dei canali ionici difettosi dalla mutazione CFTR. Ciò può migliorare la funzione polmonare del paziente, aumentare il suo peso, ridurre il contenuto di sale nel sudore e ridurre la frequenza delle infezioni polmonari.
Tuttavia, si tratta di terapie specifiche per mutazione, il che significa: i modulatori CFTR funzionano solo con determinate mutazioni e quindi solo per determinati pazienti. Qualche esempio:
Ivacaftor è stato approvato come primo modulatore CFTR nel 2012. Può essere prescritto a bambini dai 12 mesi e con un peso di almeno sette chilogrammi se hanno almeno una cosiddetta mutazione di gating (come G551D).
I pazienti con fibrosi cistica di età pari o superiore a 12 anni che hanno due copie della mutazione F508del possono essere trattati con una combinazione dei due modulatori CFTR ivacaftor e lumacaftor.
I ricercatori stanno lavorando intensamente su altri farmaci che aiuteranno contro altre mutazioni alla base della fibrosi cistica.
Terapie fisiche
Varie tecniche di terapia fisica aiutano a liberare le vie aeree dalle secrezioni bronchiali dure. Questi includono lo stoccaggio del drenaggio, il drenaggio delle vibrazioni, il massaggio con picchiettio e l'allenamento per la tosse. I genitori di bambini piccoli con fibrosi cistica possono apprendere queste tecniche di terapia respiratoria e usarle quotidianamente a casa. I bambini più grandi e gli adulti con fibrosi cistica possono eseguire la terapia respiratoria in modo indipendente (dopo essere stati istruiti da un terapista).
Trapianto di polmone - l'ultima speranza
Se la fibrosi cistica è grave, un trapianto di polmone è un'opzione. Per quanto drastico possa sembrare questo passaggio a prima vista, molti pazienti possono quindi condurre una vita con un numero significativamente inferiore di sintomi.
Mangiare correttamente per la fibrosi cistica
Poiché nella fibrosi cistica la digestione è disturbata, i pazienti devono prestare molta attenzione alla loro dieta. I pasti dovrebbero fornire abbastanza calorie e proteine per sostenere una crescita normale. Si raccomanda anche un apporto di grassi da normale a elevato. Ai pazienti vengono somministrati enzimi digestivi per supportare l'utilizzo del cibo. Esistono anche preparati con vitamine liposolubili (A, D, E, K) e minerali. Questi ultimi sostituiscono i sali che i pazienti sudano in grandi quantità.
Una certa quantità di sale da cucina dovrebbe essere assunta con il cibo durante la giornata. Corrispondentemente più è richiesto in caso di temperature elevate, febbre, diarrea, vomito. Un'eventuale carenza di ferro va testata una volta all'anno e sostituita in caso di carenza.
Neonati con fibrosi cistica
I bambini con fibrosi cistica dovrebbero essere nutriti solo con latte materno per i primi quattro mesi, purché siano prosperi. Ci sono anche prove che l'allattamento al seno per più di sei mesi è associato a una minore gravità della malattia. La ricerca ha dimostrato che i bambini allattati al seno hanno una migliore funzione polmonare e meno infezioni entro i primi tre anni di vita rispetto ai bambini con fibrosi cistica che ricevono latte artificiale.
Se l'allattamento al seno non è possibile, i lattanti dovrebbero ricevere una formula commerciale quando stanno bene.
Se i bambini con fibrosi cistica non stanno bene, si raccomanda un latte artificiale ad alto contenuto calorico (oltre al latte materno per i bambini che allattano).
Per quanto riguarda l'introduzione di alimenti complementari, valgono le stesse raccomandazioni dei bambini sani: i genitori dovrebbero iniziare l'alimentazione supplementare non prima dell'inizio del 5° mese e al più tardi all'inizio del 7° mese.
La dieta dei bambini con fibrosi cistica dovrebbe essere adattata alle loro esigenze in modo che i più piccoli possano prosperare in base alla loro età. Sia il sovrappeso che il sottopeso dovrebbero essere evitati. Si consiglia anche una dieta di buona qualità. Dovresti prestare attenzione ai grassi dietetici buoni (con acidi grassi monoinsaturi e polinsaturi, acidi grassi omega-3).
La funzione polmonare o la crescita degli alveoli è influenzata dallo sviluppo fisico nei primi anni di vita. Pertanto, i bambini con fibrosi cistica in questa fascia di età dovrebbero essere regolarmente esaminati per il loro stato nutrizionale (basato su misurazioni di peso, lunghezza del corpo e circonferenza della testa).
Fare le vaccinazioni
Le vaccinazioni sono particolarmente importanti per le persone con fibrosi cistica. I batteri se la passano più facilmente con loro e spesso sono più malati dei pazienti che non sono pre-stressati.
Fondamentalmente, i bambini con fibrosi cistica dovrebbero ricevere tutte le vaccinazioni che la Commissione vaccinale permanente (STIKO) raccomanda per la prole sana. Inoltre, si raccomanda una vaccinazione contro l'epatite A per i bambini di età pari o superiore a 12 mesi e una vaccinazione antinfluenzale annuale con il vaccino antinfluenzale inattivato tetravalente per i bambini di età pari o superiore a sei mesi. Per i bambini di età inferiore ai sei mesi, tutti i membri della famiglia e gli operatori sanitari dovrebbero essere vaccinati contro l'influenza per proteggere il bambino.
Ai bambini con fibrosi cistica può anche essere somministrato un farmaco preventivo (palivizumab) contro il patogeno RSV (virus respiratorio sinciziale). Questi virus, diffusi in tutto il mondo, possono causare malattie respiratorie, soprattutto nei neonati e nei bambini piccoli.
Fibrosi cistica: decorso della malattia e prognosi
La fibrosi cistica è causata da un cambiamento nel corredo genetico ed è quindi incurabile. In generale, l'aspettativa di vita e la qualità della vita sono di solito significativamente ridotte nella fibrosi cistica. Senza terapia, lo stato di salute si deteriora rapidamente e le persone colpite di solito non vivono a lungo.
Con una terapia tempestiva e coerente, il decorso della malattia può essere notevolmente rallentato. I pazienti ora vivono molto più a lungo rispetto a qualche anno fa. L'aspettativa di vita media con la fibrosi cistica è attualmente di circa 50 anni. Ma molti pazienti stanno anche invecchiando.
Complicazioni e malattie secondarie
Anche con una terapia intensiva, le complicazioni possono verificarsi ripetutamente con la fibrosi cistica. Molto spesso, la mancanza di respiro acuta si verifica a causa della scarsa ventilazione polmonare. Singole aree dei polmoni possono persino collassare (atelettasia).
Spesso si sviluppa bronchite cronica o polmonite. I funghi possono colpire anche i polmoni.
Inoltre, i cambiamenti nell'equilibrio di liquidi ed elettroliti possono innescare shock e insufficienza circolatoria.
Altre possibili complicanze e malattie secondarie della fibrosi cistica sono:
- malattia epatica cronica, in particolare cirrosi epatica
- Infiammazione della cistifellea e dei calcoli biliari
- pancreatite cronica
- funzione cardiaca disturbata
- ostruzione intestinale acuta (ileo)
- Invaginazione intestinale (intussuscezione)
- Malnutrizione
- Diabete mellito
- ridotta fertilità nelle donne e infertilità negli uomini
Fibrosi cistica: prevenzione?
La fibrosi cistica è una malattia ereditaria, quindi la prevenzione non è possibile. Le persone con un aumentato rischio familiare dovrebbero cercare una consulenza genetica se desiderano avere figli. Viene eseguito un test genetico per determinare se il gene CFTR è stato modificato. A seconda che uno o entrambi i partner siano portatori del gene difettoso, è possibile calcolare il rischio per la prole.
Nel frattempo, la diagnosi preimpianto (PGD) è possibile per la fibrosi cistica nella maggior parte dei paesi europei in base alle disposizioni legali del rispettivo paese. Gli ovociti vengono fecondati al di fuori dell'utero e vengono utilizzati solo embrioni senza i geni problematici della fibrosi cistica. Il prerequisito per questo è sempre l'approvazione di un comitato etico.
Informazioni aggiuntive
Linee guida:
- Linea guida di consenso S2 "Diagnosi della fibrosi cistica" della Society for Pediatric Pneumology
- Linea guida S3 "Lung disease in cystic fibrosis" della Society for Pediatric Pneumology
- Linea guida S3: "Fibrosi cistica nei bambini nei primi due anni di vita, diagnostica e terapia" della Society for Pediatric Pneumology (GPP), della German Society for Child and Adolescent Medicine (DGKJ) e altri
Gruppi di auto aiuto:
- Fibrosi cistica e.V.: https://www.muko.info/
- CF Austria: la fibrosi cistica aiuta l'Austria: https://www.cf-austria.at/
- Fibrosi cistica Svizzera: https://cystischefibroseschweiz.ch/