Sindrome di Kallmann
Astrid Leitner ha studiato medicina veterinaria a Vienna. Dopo dieci anni di pratica veterinaria e la nascita della figlia, è passata, più per caso, al giornalismo medico. Divenne subito chiaro che il suo interesse per gli argomenti medici e il suo amore per la scrittura erano la combinazione perfetta per lei. Astrid Leitner vive con figlia, cane e gatto a Vienna e nell'Alta Austria.
Maggiori informazioni sugli esperti di Tutti i contenuti di sono controllati da giornalisti medici.La sindrome di Kallmann è una malattia congenita in cui vengono prodotti pochi o nessun ormone sessuale. Le persone colpite non attraversano la pubertà; se non trattate, sia gli uomini che le donne sono sterili. Inoltre, l'olfatto è solitamente disturbato: i pazienti con KS hanno solo un odore molto limitato o per niente. Leggi tutto quello che c'è da sapere sulla “sindrome olfattogenitale” qui!
Codici ICD per questa malattia: i codici ICD sono codici riconosciuti a livello internazionale per le diagnosi mediche. Si trovano, ad esempio, nelle lettere dei medici o nei certificati di inabilità al lavoro. E23

Breve panoramica
- Cos'è la sindrome di Kallmann? Disturbo dello sviluppo congenito che porta alla mancanza di ormoni sessuali e quindi all'assenza della pubertà. Inoltre, la maggior parte dei pazienti non ha il senso dell'olfatto.
- Cause: cambiamenti genetici congeniti (mutazioni)
- Fattori di rischio: la malattia si verifica nelle famiglie in circa il 30% dei pazienti.
- Sintomi: Mancato sviluppo della pubertà (sottosviluppo del pene, dei testicoli e della prostata, poco pubico, ascelle e peli del corpo, mancanza di crescita della barba, assenza del primo periodo mestruale), infertilità, mancanza di piacere, olfatto assente o molto conseguenza ridotta e a lungo termine: osteoporosi
- Diagnostica: sintomi fisici come mancanza di sviluppo dei caratteri sessuali secondari, analisi ormonale, test genetici, ultrasuoni, tomografia a risonanza magnetica, tomografia computerizzata, raggi X
- Trattamento: terapia sostitutiva con farmaci ormonali
- Prevenzione: nessuna prevenzione possibile
Cos'è la sindrome di Kallmann?
La sindrome di Kallmann (KS, sindrome olfattogenitale, sindrome di De-Morsier-Kallmann) è un disturbo congenito dello sviluppo del cervello. Assicura che le persone colpite non producano ormoni sessuali e quindi la maturità sessuale non si verifica. Inoltre, la maggior parte dei pazienti con KS soffre di anosmia, il che significa che non percepiscono alcun odore.
La causa della malattia è un cambiamento genetico (mutazione) che colpisce già lo sviluppo embrionale nell'utero. Nel centro di controllo ormonale del cervello (ipotalamo) mancano alcune cellule responsabili del controllo prioritario della produzione di ormoni sessuali in età avanzata. Inoltre, la modificazione genetica significa che il centro olfattivo nel cervello (bulbo olfattivo) non è sviluppato o è solo incompleto. Le persone colpite non sentono alcun odore o solo in misura molto limitata.
La malattia prende il nome dallo psichiatra tedesco Franz Josef Kallmann, che la descrisse nel 1944 come "ipogonadismo (mancanza di ormoni sessuali) e anosmia (mancanza dell'olfatto)". Il termine sinonimo “Sindrome di De Morsier-Kallmann” si riferisce anche al neurologo svizzero Georges de Morsier, che ha svolto anche studi sulla sindrome di Kallmann.
Ipogonadismo secondario
La sindrome di Kallmann è una sottoforma dell'ipogonadismo secondario. Ciò significa una mancanza di ormoni sessuali, che è innescata da un malfunzionamento nel centro di controllo ormonale nel cervello (ghiandola pituitaria). Le gonadi (ovaie, testicoli) funzionano normalmente, ma ricevono pochi o nessun segnale sulla produzione di ormoni sessuali. Se c'è anosmia (mancanza dell'olfatto) oltre al deficit ormonale, i medici parlano della sindrome di Kallmann.
frequenza
La sindrome di Kallmann è una malattia rara; gli uomini ne sono colpiti più frequentemente delle donne: in media ne sono affetti circa uno su 10.000 uomini e una donna su 50.000.
Sintomi
I principali sintomi della sindrome di Kallmann sono un senso dell'olfatto notevolmente ridotto o assente e l'assenza di sviluppo puberale. La gravità dei sintomi varia da paziente a paziente. Se non trattati, entrambi i sessi rimangono sterili.
Pubertà assente: gli ormoni sessuali sono essenziali per l'inizio della pubertà. Se sono presenti troppo pochi o nessun ormone sessuale, non si verificherà la pubertà e quindi lo sviluppo dei caratteri sessuali secondari.
Sintomi nei ragazzi:
- Pochi o assenti peli pubici, ascellari e corporei
- barba mancante
- Non riuscire a rompere la sua voce
- Micropene e piccoli testicoli: nei giovani, il pene e i testicoli non si sviluppano.
- Testicoli ritenuti (spesso evidenti nell'infanzia)
- Alta statura: nelle persone sane, le placche di crescita nelle ossa lunghe si chiudono non appena viene raggiunto un determinato livello di ormoni sessuali. Se mancano gli ormoni, le persone colpite crescono in modo sproporzionato. Sono più alti dei loro genitori e hanno braccia e gambe lunghe.
Sintomi nelle ragazze:
Nelle ragazze, i sintomi sono generalmente meno pronunciati. Spesso l'unico sintomo è l'assenza della prima mestruazione (amenorrea primaria), motivo per cui la malattia viene spesso riconosciuta tardivamente. A differenza dei ragazzi, lo sviluppo fisico è in gran parte normale nonostante la malattia. Ad esempio, i seni sono generalmente sviluppati normalmente.
Sintomi negli adulti di sesso maschile:
Gli uomini con sindrome di Kallmann hanno tipicamente una ridotta densità ossea (osteoporosi) e massa muscolare. Spesso hanno un aspetto esteriore femminile dovuto a una distribuzione del grasso femminile. Hai una disfunzione erettile e sei sterile.
Olfatto ridotto o assente: i pazienti con KS non percepiscono gli odori (anosmia) o li percepiscono solo debolmente (iposmia). Questo sintomo passa spesso inosservato perché le persone sono abituate a non annusare nulla dalla nascita. La mancanza dell'olfatto può portare a situazioni pericolose nei singoli casi, ad esempio se le persone colpite non percepiscono l'odore di bruciato o l'odore di cibo avariato.
Malformazioni: altre malformazioni fisiche si verificano meno frequentemente nei pazienti con KS. Questi sono, ad esempio, disturbi dell'udito, labbro leporino, mascella e palato o denti mancanti. Alcune persone nascono con un solo rene. Lo sviluppo mentale è di solito normale in KS.
Osteoporosi: gli ormoni sessuali come il testosterone e gli estrogeni svolgono un ruolo importante nella mineralizzazione ossea. Se mancano gli ormoni, le ossa non sono così stabili come nelle persone sane - aumenta il rischio di fratture ossee.
Causa e fattori di rischio
La causa della sindrome di Kallmann è un cambiamento genetico innato (mutazione).Di solito nasce spontaneamente, in circa il 30 per cento dei casi viene trasmesso alla prole da uno o entrambi i genitori.
Il cambiamento genetico colpisce già lo sviluppo embrionale nell'utero: da cellule progenitrici comuni nascono le cellule dell'olfatto e quelle preposte al controllo delle gonadi (ovaie, testicoli).
Nella sindrome di Kallmann, lo sviluppo di queste cellule progenitrici è disturbato dal cambiamento genetico. Di conseguenza, le cellule responsabili del controllo prioritario della produzione di ormoni sessuali e le cellule olfattive non sono sufficientemente sviluppate. Le persone affette non sviluppano la pubertà e non percepiscono alcun odore.
Ormoni sessuali
L'ipotalamo (una regione del diencefalo) è il centro di controllo ormonale del corpo. Nelle persone sane, l'ipotalamo rilascia l'ormone GnRH (ormone di rilascio della gonadotropina). Questo a sua volta stimola la ghiandola pituitaria a rilasciare gli ormoni LH (ormone luteinizzante) e FSH (ormone follicolo-stimolante).
LH e FSH agiscono sulle gonadi (ovaie, testicoli), che alla fine producono gli ormoni sessuali: nelle donne, gli ormoni sessuali femminili estrogeni e progesterone promuovono la maturazione degli ovociti e innescano l'ovulazione, negli uomini l'ormone testosterone provoca la formazione di sperma.
Durante la pubertà inizia la produzione di ormoni sessuali e con essa la maturazione sessuale. Se, come nella sindrome di Kallmann, c'è poco o nessun GnRH presente, vengono prodotti troppo poco o nessun ormone sessuale e la pubertà non si sviluppa. Se non trattate, le persone colpite non raggiungono la maturità sessuale e rimangono sterili.
Fattori di rischio
Nella maggior parte dei casi, il cambiamento genetico avviene spontaneamente. Come questo avvenga non è chiaro. In circa il 30 per cento di tutti i casi, la malattia si manifesta nelle famiglie: le persone colpite hanno ereditato la mutazione da uno o entrambi i genitori.
Finora sono state descritte diverse mutazioni che causano la sindrome di Kallmann. Questi includono le mutazioni con i nomi KAL1, FGFR1, FGF8, CHD7, SOX10, PROKR2 e PROK2.
Indagine e diagnosi
La diagnosi della sindrome di Kallmann è in molti casi tardiva, poiché i sintomi spesso si manifestano solo nell'adolescenza. Questo vale soprattutto per i ragazzi, nei quali la mancanza di sviluppo sessuale è solitamente più chiaramente visibile. Nelle ragazze i sintomi sono spesso meno pronunciati, tanto che solo l'assenza del primo ciclo mestruale dà luogo a una visita medica. Inoltre, in molti casi le persone colpite non sospettano una malattia dietro i sintomi, ma credono piuttosto che siano "a sviluppo tardivo".
In alcuni casi, la malattia è già evidente nei neonati: i ragazzi affetti possono avere testicoli ritenuti (criptorchidismo) e/o un pene molto piccolo (micropene).
Il primo punto di contatto in caso di segni di sindrome di KS è il pediatra, in caso di involontaria infanzia il ginecologo o l'urologo.
Il medico effettua i seguenti esami:
Storia familiare: se si sospetta la sindrome di Kallmann, il medico chiede se ci sono casi noti di KS in famiglia.
Esame obiettivo: caratteristiche sessuali secondarie sottosviluppate come un pene troppo piccolo per la sua età o l'assenza del primo periodo mestruale danno al medico i primi indizi sulla sindrome di KS. Presta attenzione anche alle dimensioni del corpo, all'ascella, al petto e ai peli pubici e valuta la crescita della barba.
Test dell'olfatto: questo test è possibile a partire dai cinque anni circa. Il medico utilizza fragranze pure come la vanillina per verificare se il paziente può percepire l'odore.
Esame del sangue: il medico determinerà un livello ormonale modificato con un esame del sangue. Tipicamente i livelli di GnRH, LH e FSH sono diminuiti o ad un livello basso-normale. I livelli di ormoni sessuali nei ragazzi e nelle ragazze nell'adolescenza sono pre-pubescenti.
Esame ecografico dei testicoli: il medico utilizza gli ultrasuoni per esaminare i tessuti molli come i testicoli.
Tomografia computerizzata (TC) o tomografia a risonanza magnetica (MRT): con queste procedure di esame, il medico controlla se il centro olfattivo nel cervello (bulbo olfattivo) è sviluppato.
Esame radiografico della mano: Con l'aiuto di un esame radiografico della mano, il medico determina se le cartilagini di accrescimento sono già chiuse e il corpo ha quindi terminato la crescita.
Spermiogramma: il medico esamina se ci sono spermatozoi nell'eiaculato.
Test genetico: il test genetico viene utilizzato per determinare l'esatta mutazione che causa la malattia.
Come viene trattata la sindrome di Kallmann?
La sindrome di Kallmann è facilmente curabile. Le persone colpite ricevono un trattamento sostitutivo con ormoni sessuali. Se la malattia viene riconosciuta e trattata prima della pubertà, le persone colpite di solito conducono una vita in gran parte senza restrizioni. Ciò include lo sviluppo regolare della pubertà e una vita sessuale normale.
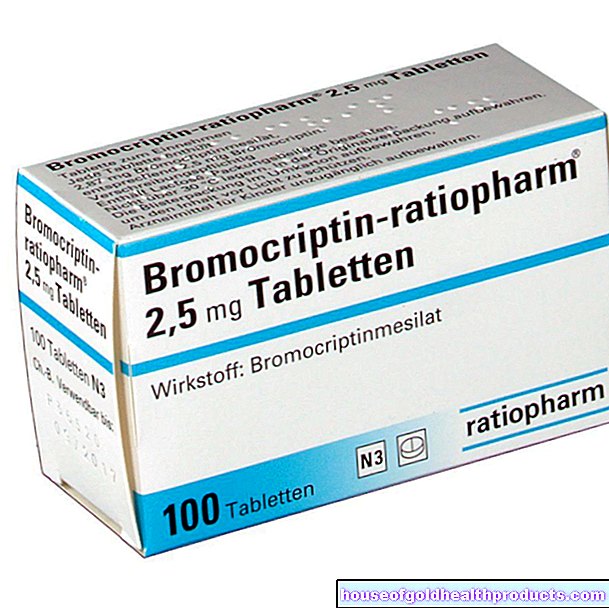
Somministrazione degli ormoni sessuali: gli uomini ricevono testosterone, le donne estrogeni e progesterone. I preparati ormonali sono disponibili sotto forma di iniezioni, gel o cerotti. La terapia ormonale è di solito continuata per tutta la vita negli uomini e fino alla menopausa nelle donne.
Nel 10-20% dei casi, una carenza congenita di GnRH si risolve al termine della terapia ormonale sostitutiva. Dopo la terapia, i pazienti hanno livelli ormonali normali e sperimentano la normale maturità sessuale. Per questo motivo, i medici raccomandano di sospendere la terapia ogni uno o due anni per determinare la necessità di continuare la terapia.
Somministrare ormoni sessuali se si desidera avere figli: il corpo ha bisogno di GnRH per la formazione dello sperma. Per questo motivo, agli uomini che vogliono avere un figlio viene somministrato l'ormone GnRH invece del testosterone. Ci vogliono dai 18 ai 24 mesi per iniziare la produzione di sperma. In circa l'80% dei casi, gli uomini sono fertili in seguito. Gli uomini con testicoli ritenuti hanno una prognosi leggermente meno favorevole.
Trattamento dell'osteoporosi: i pazienti con ridotta densità ossea ricevono calcio e vitamina D. Inoltre, si raccomandano sport ed esercizio fisico per rafforzare le ossa.
Anosmia: Al momento non esiste una terapia per ripristinare l'olfatto.
Psicoterapia: per alcuni pazienti con KS, la malattia è un grave onere psicologico. Lo psicoterapeuta è il contatto giusto qui.
Decorso della malattia e prognosi
Il modo in cui progredisce la sindrome di Kallmann varia da persona a persona. I sintomi variano da paziente a paziente. È possibile che la malattia si manifesti già nell'infanzia, diagnosticata nell'adolescenza o che il deficit ormonale compaia solo nell'età adulta.
Se la malattia viene diagnosticata in tempo utile, la prognosi è molto buona. La maturazione sessuale è raggiunta in tutti i pazienti con un appropriato trattamento ormonale. I pazienti sperimentano uno sviluppo regolare della pubertà, sono fertili e hanno un'aspettativa di vita normale. Quasi tutti i pazienti che desiderano avere figli diventano fertili con un trattamento appropriato.
Se KS viene riconosciuto e trattato solo dopo i 16 anni, potrebbe essere già diventato alto. Questo cambiamento non può essere annullato nemmeno con i farmaci.
Prevenire
Poiché si tratta di una malattia genetica, non è possibile alcuna prevenzione.
Tags.: notizia prevenzione gpp